

Sau Hướng dẫn cập nhật về hội chứng Brugada năm 2013 của các Hội chuyên ngành lớn trên thế giới, về chẩn đoán hội chứng Brugada có nhiều điểm đơn giản đi, nhưng thực tế lại gặp nhiều trở ngại do đơn giản.
TS Phạm Hữu Văn
Từ thực tế, cần phải có tiêu chuẩn chặt chẽ hơn cũng như thống nhất nhiều danh pháp về hội chứng sóng J bao gồm tái cực sớm và hội chứng Brugada. Chúng tôi xin giới thiệu Bản đồng thuận về hội chứng sóng J với mong muốn cung cấp cho các đồng nghiệp những cập nhật mới nhất về vấn đề này.
Hướng dẫn đồng thuận này được các Hội Nhịp tim Châu Á Thái Bình Dương (Asia Pacific Heart Rhythm Society: APHRS), Hội Nhịp Tim châu Âu (European Heart Rhythm Association: EHRA), Hội Nhịp Tim (Heart Rhythm Society: HRS), Hội Tạo nhịp Tim và Điện sinh lý Mỹ La tinh (Latin American Society of Cardiac Pacing and Electrophysiology) thông qua và xuất bản ngày 13/07/2016.
Các từ quan trọng
Đột tử tim (Sudden cardiac death); sóng J (J wave); Hội chứng Brugada (Brugada syndrome); Hội chứng tái cực sớm (Early repolarization syndrome); Rối loạn nhịp tim (Cardiac arrhythmia); Rung thất (Ventricular fibrillation); Hội chứng rối loạn nhịp tim di truyền (Inherited cardiac arrhythmia syndrome).
Lời mở đầu
Các hội chứng sóng J (J-wave syndromes: JWSs), gồm hội chứng Brugada (BrS) và hội chứng tái cực sớm (ERS), đã thu hút sự quan tâm của cộng đồng tim mạch học trong hơn 2 thập kỷ qua sau khi xác định BrS là một thực thể lâm sàng mới do Pedro và Josep Brugada đưa ra vào năm 1992. [1] Tác động lâm sàng của ERS chưa được đánh giá đầy đủ cho đến tận năm 2008. [2 – 4] Các hội nghị đồng thuận dành cho BrS đã được tổ chức vào năm 2000 và năm 2004 [5,6], nhưng một hội nghị đồng thuận đặc biệt tập trung vào ERS chưa được nhóm họp trước đây ngoại trừ bàn về thuật ngữ, cũng như các hướng dẫn cho cả hai hội chứng đã được xem xét lần cuối cùng vào năm 2013. [7] Rất nhiều thông tin mới đã xuất hiện từ đó. Diễn đàn hiện nay được tổ chức để đánh giá các thông tin mới và nêu bật các khái niệm mới về chẩn đoán phân biệt, tiên lượng, cơ chế tế bào và ion, cũng như cách tiếp cận điều trị JWSs. Các chuyên gia hàng đầu, bao gồm các thành viên của Hội Nhịp tim (HRS), Hội Nhịp Tim Châu Âu (EHRA), Hội Nhịp Tim Châu Á Thái Bình Dương (APHRS), đã họp tại Thượng Hải, Trung Quốc vào tháng 4 năm 2015. Nhóm Công tác với việc xem xét các khái niệm mới và đánh giá các bằng chứng mới cho hoặc là chống lại các thủ thuật chẩn đoán và điều trị riêng biệt. Mọi nỗ lực đã được thực hiện để tránh bất kỳ xung đột lợi ích thực tế, tiềm ẩn hoặc cảm nhận nào có thể nảy sinh do mối quan hệ bên ngoài hoặc lợi ích cá nhân. Báo cáo đồng thuận này nhằm hỗ trợ các nhà cung cấp dịch vụ chăm sóc sức khoẻ trong việc ra quyết định lâm sàng. Tuy nhiên, phán quyết cuối cùng về chăm sóc một bệnh nhân cụ thể phải do nhà cung cấp dịch vụ chăm sóc sức khoẻ thực hiện dựa trên tất cả các sự kiện và hoàn cảnh bệnh nhân biểu hiện.
Các thành viên của nhóm Công tác này được lựa chọn để đại diện cho các chuyên gia tham gia vào việc chăm sóc y tế cho bệnh nhân với JWS, cũng như những người tham gia nghiên cứu về các cơ chế gây ra các hội chứng này. Các chuyên gia được lựa chọn trong lĩnh vực này đã tiến hành rà soát toàn diện y văn. Đánh giá các phương pháp chẩn đoán, phân tầng nguy cơ, cách tiếp cận điều trị, cũng như nhìn nhận sâu sắc hơn về cơ chế được thực hiện, bao gồm đánh giá tỷ lệ nguy cơ /lợi ích. Mức độ bằng chứng và sức mạnh của việc khuyến cáo các lựa chọn điều chỉnh riêng biệt được cân nhắc và phân loại. Các khuyến cáo với các chỉ dẫn class được lấy từ các tuyên bố hoặc hướng dẫn của HRS, EHRA, APHRS và/hoặc ESC. [8,9] Các khuyến cáo không có chỉ định class được lấy từ sự đồng thuận nhất trí của các tác giả. Các khuyến nghị đồng thuận trong tài liệu này sử dụng các phân loại Class I, IIa, IIb và III thường được sử dụng và ngôn ngữ tương ứng: “được đề nghị” cho một khuyến cáo đồng thuận class I; “có thể hữu ích” hoặc “hợp lý” đối với khuyến cáo đồng thuận của class IIa; “có thể được xem xét” đối với khuyến cáo đồng thuận của class IIb; và “không được khuyến khích” đối với khuyến cáo đồng thuận class III.
Giới thiệu
Sự xuất hiện của các sóng J chiếm ưu thế trên điện tâm đồ (ECG) đã được báo cáo từ lâu trong các trường hợp hạ thân nhiệt [10-12] và tăng kali máu [13,14]. Gần đây, sóng J đã được nhấn mạnh liên kết với rối loạn nhịp thất. [15] Trong các tình huống này, sóng J điển hình có thể có bề rộng và cao khi suất hiện đoạn ST chênh lên, như ở các trường hợp BrS. Ở người, sóng J bình thường thường xuất hiện khi chênh lên của đoạn ST, với một phần của sóng J được xen vào bên trong QRS. Một mẫu tái cực sớm (ERP) trong ECG, bao gồm một sóng J riêng biệt hoặc điểm J chênh lên, hoặc sự chễ đôi (notch) hoặc líu díu (slur) ở phần đầu cuối của QRS có và không có sự chênh lên của đoạn ST, theo truyền thống được xem là lành tính. [16,17] Đặc tính lành tính của ERP đã được thử thách vào năm 2000.[18] Dựa trên số liệu thực nghiệm cho thấy biểu hiện ECG này có xu hướng phát triển nhịp nhanh thất đa hình (VT) và rung thất (VF) trong các can thiệp tưới máu mạch vành.[15,18 – 20] Xác nhận giả thuyết này đã được Haissaguerre và cộng sự,[2] Nam và cộng sự,[3] và Rosso cùng cộng sự đưa ra 8 năm sau. Các nghiên cứu trên cùng với các nghiên cứu dựa trên dân số khác đã cung cấp bằng chứng lâm sàng về nguy cơ gia tăng phát triển các biến cố loạn nhịp đe dọa tính mạng và tử vong đột ngột do tim (SCD) trong số các bệnh nhân có biểu hiện ERP, đặc biệt ở các chuyển đạo dưới và bên – dưới. Sự thiếu đồng thuận về thuật ngữ liên quan đến tái cực sớm (ER) đã dẫn tới nhiều nhầm lẫn và không nhất quán trong các báo cáo [21 – 23] Một báo cáo thống nhất gần đây của chuyên gia tập trung vào các thuật ngữ của ER khuyến cáo chóp (peak) của kết thúc QRS chẽ đôi (notch) và/hoặc khởi đầu (onset) của kết thúc QRS líu díu (slur) được gọi như là Jp và Jp cần vượt quá 0,1 mV ở ≥ 2 chuyển đạo dưới và/hoặc bên liên tiếp của ECG 12 chuyển đạo chuẩn đối với biểu hiện ER [24]. Nó đã được khuyến cáo thêm sự bắt đầu của kết thúc QRS chẽ đôi hoặc sóng J được gọi như Jo và kết thúc như Jt.
ERS và BrS được cho là đại diện cho 2 biểu hiện của JWS. Cả hai hội chứng này liên quan đến khả năng dễ bị tổn thương do sự phát triển VT đa hình và VF dẫn đến SCD [1-3,5] ở người trưởng thành trẻ tuổi mà không có bệnh tim cấu trúc rõ ràng và đôi khi là hội chứng đột tử ở trẻ sơ sinh.[25-27] Khu vực thường bị ảnh hưởng nhiều nhất trong BrS là đường ra thất phải (RVOT); Trong ERS, khu vực này là vùng dưới của thất trái (LV).[2,4,28 – 32] Do đó, BrS được đặc trưng bằng các sóng J chiến ưu thế biểu hiện như sự chênh lên của đoạn ST type vòm ở các chuyển đạo trước tim phải V1 -V3, trong khi ERS có đặc điểm sóng J, Jo chênh lên, chẽ đôi hoặc líu díu của phần kết thúc của QRS, đoạn ST hoặc Jt chênh lên ở chuyển đạo bên (type 1), bên dưới (type 2), hoặc dưới bên cộng với trước hoặc chuyển đạo thất phải (RV) (type 3).[15] ERP thường thấy ở những cá thể khoẻ mạnh, đặc biệt là ở nam thanh niên, cá nhân da đen và vận động viên. ERP cũng được quan sát thấy trong các điều kiện mắc phải, gồm hạ thân nhiệt và thiếu máu cục bộ.[15,33,34] Khi kết hợp với VT/VF khi không có bệnh tim, mẫu tái cực sớm (ERP) được gọi là hội chứng tái cực sớm (ERS).
Tỷ lệ hiện mắc của BrS với ECG type 1 ở người lớn ở các nước châu Á cao hơn ở các nước châu Á, như Nhật Bản (0,15% – 0,27%) [35,36] và Phi-líp-pin (0,18%), [37] và ở người Mỹ gốc Nhật ở Bắc Mỹ ( 0,15%) [38] so với các nước phương Tây, bao gồm châu Âu (0% – 0,017%) [39 – 41] và Bắc Mỹ (0,005% -0,1%). [42,43] Ngược lại, tỷ lệ ERP ở các chuyển đạo dưới và / hoặc bên với sự chênh lên của điểm J ≥ 0,1 mV dao động từ 1% đến 24% và đối với sự chênh lên của điểm J > 0,2 mV dao động từ 0,6% đến 6,4% [44-46]. Không có sự khác biệt khu vực có ý nghĩa trong tỷ lệ ERP đã được thông báo.[47] Tuy nhiên, ERP phổ biến đáng kể hơn ở người da đen so với người ở da trắng gốc đông nam châu Âu (Caucasians). Rất ít báo cáo về sự khác biệt giữa các vùng trong việc thể hiện ERS. ERP xuất hiện phổ biến hơn ở người Úc gốc Thổ Dân so với người Úc Da trắng.[48]
Cập nhật về chẩn đoán BrS
Theo báo cáo đồng thuận năm 2013 về rối loạn nhịp tim di truyền [8] và hướng dẫn năm 2015 cho điều chỉnh các bệnh nhân rối loạn nhịp thất và ngăn ngừa SCD [9] : “BrS được chẩn đoán ở bệnh nhân có chênh lên của ST với hình thái type 1 ≥2 mm ở ≥ 1 chuyển đạo trong số các chuyển đạo trước tim V1, V2, đặt ở các khoảng liên sườn thứ 2, 3 hoặc 4 xuất hiện hoặc tự phát hoặc sau test thuốc thúc đẩy bằng sử dụng các thuốc chống rối loạn nhịp Class I. BrS được chẩn đoán ở bệnh nhân có type 2 hoặc 3 với đoạn ST chênh lên ≥1 chuyển đạo trong số các chuyển đạo trước tim phải V1, V2 ở khoảng liên sườn thứ 2, thứ 3 hoặc thứ 4 khi test thuốc thúc đẩy bằng đường tĩnh mạch gồm các thuốc chống loạn nhịp Class I tạo ra hình thái ECG type I”.
Nhóm công tác hiện nay lo ngại điều này có thể dẫn đến chẩn đoán BrS quá mức, đặc biệt ở những bệnh nhân biểu hiện ECG type 1 sau test thử bằng thuốc. Dữ liệu cho thấy dân số sau này có nguy cơ rất thấp và tỷ lệ dương tính giả được giả định do thử bằng thuốc không phải không đáng kể. Mặc dù đã tiến hành một quy trình nghiêm ngặt để đưa ra các hướng dẫn trước nhưng vẫn không có tiêu chuẩn vàng để xác định chẩn đoán, đặc biệt ở những bệnh nhân có ít bằng chứng về bệnh. Theo đó, người ta khuyến cáo nên áp dụng các tiêu chuẩn chẩn đoán và hệ thống điểm số cho BrS. Phù hợp với khuyến cáo của các hướng dẫn năm 2013 và 2015, chỉ có ST chênh lên type 1 (“kiểu vòm”) được xem xét chẩn đoán BrS (Hình 1) và BrS có đặc điểm là đoạn ST chênh lên > 2 mm (0,2 mV) trong ≥ 1 chuyển đạo trước tim phải (V1 -V3) ở khoang liên sườn thứ 4, 3, hoặc 2. Tuy nhiên, như một sự khởi đầu từ hướng dẫn, báo cáo đồng thuận khuyến cáo khi đoạn ST chênh lên type 1 được bộc lộ bằng sử dụng block kênh natri (Bảng 1), chẩn đoán BrS cần yêu cầu bệnh nhân cũng có một trong các điều sau đây: VF hoặc VT đa hình, ngất có thể do nguyên nhân rối loạn nhịp tim, tiền sử gia đình có SCD ở tuổi < 45 với khám nghiệm tử thi âm tính, ECG kiểu vòm trong các thành viên trong gia đình, hoặc hô hấp ngắc ngoải ban đêm, có khả năng tạo ra VT/VF với các nhát bóp sớm 1 hoặc 2 giúp chẩn đoán BrS trong những trường hợp này. [50]
Type 2 (“type yên ngựa”) hoặc type 3 ST chênh lên không thể thay thế type 1, ngoại trừ chuyển sang type 1 với sốt hoặc thử thuốc chẹn kênh natri. Thử thuốc – type 1 được tạo ra có thể được sử dụng để chẩn đoán BrS chỉ khi được kèm theo một trong các tiêu chí nêu trên. Loại 2 có đặc điểm là chênh lên của đoạn ST ≥ 0,5 mm (thường ≥2 mm ở V2) ở ≥ 1 chuyển đạo trước tim phải (V1 -V3), tiếp theo là ST lồi lên. Đoạn ST tiếp theo là sóng T dương ở V2 và hình thái có thể thay đổi V1. Type 3 được đặc trưng bằng một hình lưng ngựa hoặc vòm với đoạn ST chênh lên < 1mm. Vị trí của các chuyển đạo trước tim phải ở các vị tri trên hơn (ở các khoang liên sườn thứ 3 hoặc thứ 2) trên ECG 12 chuyển đạo lúc nghỉ hoặc ECG Holter 12 chuyển đạo làm tăng độ nhạy cảm của ECG. [51-53] Người ta khuyến cáo ghi ECG được thực hiện ở các vị trí chuẩn và vị trí cao hơn đối với các chuyển đạo V1 và V2. Veltman và cộng sự [54] cho thấy việc định vị RVOT bằng chụp cộng hưởng từ (MRI) tương quan với sự chênh lên của đoạn ST type 1 trong BrS và vị trí chuyển đạo theo khu vực RVOT cải thiện chẩn đoán BrS. Điều thú vị là, trong hầu hết các trường hợp, mẫu type 1 được xác định ở khoang liên sườn 3 ở vị trí xương ức và cạnh ức trái.[54] Trong việc xem xét ECGs của một thuần tập lớn bệnh nhân BrS, Richter và cộng sự[55]kết luận, chuyển đạo V3 không mang lại thông tin chẩn đoán trong BrS.
Hệ thống thang điểm chẩn đoán BrS được đề xuất, được gọi là Thang điểm BrS Thượng Hải, được trình bày ở Bảng 2. Các khuyến cáo này dựa trên tài liệu sẵn có và kinh nghiệm lâm sàng của các thành viên Nhóm Chuyên trách. [8; 56 – 60]Trọng số của các biến dựa trên ý kiến chuyên gia từ các nghiên cứu thuần tập được thông báo thường không bao gồm tất cả các biến được trình bày. Do đó, các hệ số trọng lượng nghiêm ngặt, khách quan không phải là nguồn gốc từ các yếu tố nguy cơ quy mô lớn và các tập dữ liệu kết quả. Tuy nhiên, các tác giả tin rằng một số trọng số suy diễn có thể có lợi khi áp dụng cho bệnh nhân. Như với tất cả các khuyến cáo như vậy, chúng sẽ cần phải trải qua quá trình xác nhận ban đầu và đang diễn ra trong các nghiên cứu trong tương lai.
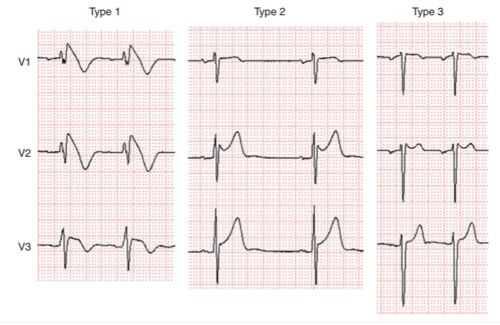
Hình 1. Ba type ST chênh lên kết hợp với Hội chứng Brugada. Chỉ có type 1 là chẩn đoán hội chứng Brugada.
Bảng 1Các thuốc được sử dụng để làm bộc lộ ECG Brugada
|
Thuốc |
Liều |
Dường sử dụng |
|
Ajmaline |
1 mg/kg over 10 minutes |
Tĩnh mạch |
|
Flecainide |
2 mg/kg over 10 minutes 200–300 mg |
Tĩnh mạch Uống (41 hour) |
|
Procainamide |
10 mg/kg over 10 minutes |
Tĩnh mạch |
|
Pilsicainide |
1 mg/kg over 10 minutes |
Tĩnh mạch |
Bảng2: Hệ thống thang điểm Thượng Hải được để nghị cho chẩn đoán hội chứng Brugada
|
|
Điểm |
|
I. ECG (12- chuyển đạo / Lưu động) |
|
|
A. Mẫu type 1 Brugada tự phát ở chuyển đạo bình thường hoặc cao |
3,5 |
|
B. Mẫu ECG type 1 Brugada được tạo ra do sốt ở các chuyển đạo bình thường hoặc cao |
3 |
|
C. Mẫu ECG type 2 hoặc 3 Brugada chuyển đổi với thử thách thuốc thúc đẩy |
2 |
|
* Chỉ có điểm thưởng một lần cho điểm số cao nhất trong phân tầng này. Một mục từ phân tầng này phải được áp dụng. |
|
|
II. Bệnh sử lâm sàng* |
|
|
A. Ngừng tim không giải thích được hoặc VF /VT đa hình được chứng minh bằng tư liệu. |
3 |
|
B. Thở ngắc ngoải về ban đêm (Nocturnal agonal respirations) |
2 |
|
C. Nghi ngờ ngất do loạn nhịp |
2 |
|
D. Ngất có cơ chế / bệnh căn không rõ |
1 |
|
E. Cuồng nhĩ / rung nhĩ ở các bệnh nhân, 30 tuổi không có căn nguyên lựa chọn. |
0,5 |
|
* Chỉ có điểm thưởng một lần cho điểm số cao nhất trong phân tầng này. |
|
|
III. Bệnh sử gia đình |
|
|
A. Thế hệ thứ nhất và thứ hai được xác định BrS |
2 |
|
B. Nghi ngờ SCD (sốt, đêm, các chất gây nghiện Brugada) ở thế hệ thứ nhất và hai. |
1 |
|
C. SCD không giải thích được, người thân thế hệ thứ nhất và hai với autopsy âm tính. |
0,5 |
|
* Chỉ có điểm thưởng một lần cho điểm số cao nhất trong phân tầng này. |
|
|
IV. Kết quả Test di truyền |
|
|
A. Đột biến bệnh lý có thể trong gene nhạy cảm Brugada. |
0,5 |
|
Điểm số (đòi hỏi phải có ít nhất 1 lần xác định ECG) |
|
|
≥3.5 điểm: Có thể / xác định BrS |
|
|
2–3 điểm: có khả năng BrS |
|
|
< 2 điểm: Không chẩn đoán |
|
BrS = Hội chứng Brugada; SCD = đột tử do tim; VF = Rung thất; VT = nhanh thất.
Test thuốc và các công cụ chẩn đoán khác
Khi có nghi ngờ lâm sàng về BrS không có chênh lên đoạn ST type 1 tự phát, thử thuốc sử dụng block kênh natri được khuyến cáo. Một danh sách các thuốc được sử dụng cho mục đích này được trình bày trong Bảng 1 (xem thêm brugadadrugs.org). Test chỉ được xem là dương tính nếu đạt được mẫu ECG type 1, và nên ngưng sử dụng trong trường hợp các ngoại tâm thất nhiều hoặc các rối loạn nhịp tim khác, hoặc QRS giãn rộng [41] 30% so với giá trị ban đầu.[6] Như một sự thay thế, “Test đổ đầy dạ dầy” đã được đề xuất cho chuẩn đoán BrS.[61] Trong trường hợp này, ECG được thực hiện trước và sau một bữa ăn lớn. Việc sử dụng “điện cực cao” làm tăng độ nhạy cảm ghi nhận sự chênh lên của đoạn ST type 1 tự phát vào ban đêm hoặc sau bữa ăn nhiều.[62] ST chênh lên type 1 được ghi nhận khi sử dụng Holter là type 1 tự phát, nó là phù hợp để giả định type 1 tự phát được ghi nhận bằng Holter về ban đêm hoặc sau bữa ăn nhiều có giá trị hơn – cho cả hai về chẩn đoán và tiên lượng – so với type 1 được tạo ra do thuốc.
Thử thuốc không được chỉ định ở những bệnh nhân không có triệu chứng biểu hiện ECG type 1 dưới điều kiện cơ bản do thiếu giá trị chẩn đoán bổ sung. Những test thuốc thúc đẩy này cũng không được khuyến cáo trong những trường hợp sốt thúc đẩy ECG type 1 đã được chứng minh bằng tư liệu, ngoại trừ mục đích nghiên cứu. Nhiều cuộc tranh luận đã tập trung vào việc xác định một thử block kênh natri dương tính giả.[63] Sự đồng thuận về dương tính giả khó để xác định do thiếu tiêu chuẩn vàng. Sự phát triển chênh lên đoạn ST type 1 trong đáp ứng với phép thử block natri nên được xem xét như tính xác suất, chứ không phải là nhị phân, trong tự nhiên. Như sẽ được thảo luận sau, một cách tiếp cận tương tự được khuyến cáo trong việc đánh giá khả năng của các biến thể di truyền để thúc đẩy các kiểu hình BrS.
Những bệnh nhân không có triệu chứng có tiền sử gia đình BrS hoặc SCD nên được thông báo về thử nghiệm thử thách của kênh natri để cung cấp một chẩn đoán chính xác hơn về BrS. Tuy nhiên, bệnh nhân nên được khuyên không nên điều trị được khuyến cáo bất kể kết cục nào do nguy cơ dài hạn của các bệnh nhân BrS được chẩn đoán bằng test này có ý nghĩa thấp hơn nguy cơ các bệnh nhân với type 1 tự phát. Các bệnh nhân cũng cần được thông báo về nguy cơ của test và về hậu quả tình thần của việc có kết quả xét nghiệm dương tính không kèm theo bằng điều trị xác định. Quyết định về việc liệu có nên thực hiện thử thuốc cuối cùng hay không nên để lại cho bệnh nhân những thông tin tốt.
Thực hiện test ajmaline ở trẻ em là vấn đề vì 2 lý do. Thứ nhất, test dường như ít nhạy cảm hơn ở trẻ em so với ở người lớn. Trong thực tế, trong 1 nghiên cứu, test ajmaline lặp lại được thực hiện sau khi tuổi dậy thì phát hiện BrS ở 23% người thân với test thuốc âm tính trước đó được thực hiện khi còn nhỏ.[64] Thứ hai, test được kết hợp với nguy cơ cao hơn so với người lớn. Trong một loạt, 10% trẻ em trải qua test ajmaline, bao gồm 3% nhóm không có triệu chứng, đã phát triển VT dai dẳng [64,65]. Cần thận trọng khi thực hiện test thử với block natri ở người lớn với đột biến kênh natri bệnh lý đã biết hoặc ở các bệnh nhân có khoảng PR kéo dài, chú ý đến người mang các đột biến như vậy.[66]
Chẩn đoán phân biệt
Các nguyên nhân khác của ST chênh lên nên được loại trừ trước khi thiết lập chẩn đoán BrS (Bảng 3). Các hình giả thứ phát do qua lọc thấp cần được loại trừ. [67]
Các tình huống tạo ra type 1 ECG giống Brugada gồm block nhánh bó phải (RBBB), các bất thường lồng ngực (pectus excavatum), bệnh cơ tim thất phải gây loạn nhịp (ARVC) và tắc động mạch vành trái hoặc nhánh conus của động mạch vành phải, cung cấp máu cho RVOT (Bảng 3A).
Sự phân biệt giữa BrS và ARVC là một thách thức đặc biệt. Mặc dù sự tranh luận về mức độ bất thường cấu trúc có trong BrS vẫn tiếp tục, hầu hết các nhà nghiên cứu đều coi BrS là bệnh kênh. Các dấu hiệu bất thường về cấu trúc, như sự xơ cơ tim về mặt tổ chức học của RVOT, nó có thể không trở nên bằng chứng khi sử dụng các kỹ thuật hình ảnh kinh điển, đã được đề xuất để tính toán hoặc bổ xung với dẫn truyền chậm và rối loạn nhịp thất trong BrS. MRI và các nghiên cứu chụp cắt lớp điện toán chùm electron ở bệnh nhân BrS đều cho thấy sự bất tường tinh tế, gồm các bất thường chuyển động thành và chức năng co bóp giảm của RV, cũng như phạm vi ít hơn của LV, và giãn RVOT .[68 – 71] Trong nghiên cứu duy nhất phân biệt giữa bệnh nhân có và không có đột biến SCN5A, không có sự khác biệt đã được quan sát thấy ở kích thước RVOT hoặc phân suất tống máu RV giữa những bệnh nhân này. Sự suy giảm lớn hơn của kích thước và phân suất tống máu đã được quan sát thấy ở những bệnh nhân có đột biến SCN5A. Sự khác biệt đáng kể đã được quan sát thấy ở kích thước RV và LV và phân suất tống máu so với những người khỏe mạnh.[72] Giãn tim và co bóp giảm ở tất cả các nghiên cứu này đã bổ xung vào sự thay đổi cấu trúc (xơ hóa, thoái hóa mỡ). Tuy nhiên, theo nhận xét của Van Hoorn và cộng sự, [72] hầu như không có dấu hiệu xơ hóa hoặc thoái hóa mỡ có thể được phát hiện, có lẽ vì độ phân giải không gian của hình ảnh được sử dụng quá thấp để phát hiện những thay đổi tinh tế như vậy.
Bảng 3.Chẩn đoán phân biệt và các yếu tố điều biến trong hội chứng Brugada
|
A. Chẩn đoán phân biệt |
B. Các yếu tố điều biến |
|
† Block nhánh bố phải không điển hình † Phì đại thất † Tái cực sớm (đặc biệt ở vận động viên) † Viêm màng ngoài tim / viêm cơ tim cấp † Thiếu máu cơ tim cục bộ hoặc nhồi máu cơ tim cấp (đặc biệt thất phải) † Thuyên tắc huyết khối phổi † Đau thắt ngực Prinzmetal † Bóc tách phình động mạch chủ † Các bất thường hệ thần kinh trung ương và tự động † Loạn dưỡng cơ Duchenne † Thất điểu Friedreich † Teo cơ Spinobulbar † Loạn dưỡng cơ † Loạn sản thất phải gây loạn nhịp † Chèn ép cơ học đường ra thất phỉ (như, biến dạng lồng ngực, u trung thất, tràn máu màng ngoài tim. † Hạ thân nhiệt † ECG sau khử rung tim |
† Các bất thường điện giải: ○ Tăng kali máu ○ Hạ kali máu ○ Tăng canxi máu ○ Hạ natri máu † Nhiệt độ: tăng thân nhiệt (sốt), hạ thân nhiệt. † Tăng testosterone máu † Điều trị với: ○ Các thuốc chống loạn nhịp: các chẹn kênh natri (Class IC, Class IA), kháng canxi, beta-blockers ○ Các thuốc chống đau thắt ngực: Kháng canxi, nitrates, các mở kênh kali ○ Các thuốc hướng tâm thần: các chống trầm cảm tricyclic/ tetracyclic, phenothiazines, ức chế tái hấp thu serotonin chọn lọc, lithium, benzodiazepines ○ Các thuốc mê /giảm đau: propofol, bupivacaine, procaine ○ Các thuốc khác: kháng histamine H1, ngộ độc rươu, cocaine, cannabis, ergonovine |
Antzelevitch và các cộng sự từ lâu đã gợi ý một cách giải thích khác [31,73,74]. Mất điện thế hoạt động (AP), đã được thấy trong các mô hình thực nghiệm để tạo ra nền loạn nhịp trong BrS, dẫn tới những thay đổi co bóp có thể giải thích các bất thường chuyển động thành được quan sát. Tái cực tất cả hay là không ở kết thúc pha 1 của điện thế hoạt động thượng tâm mạc giải thích mất đi vòm làm cho kênh caxi bất hoạt rất sớm ngay sau khi nó hoạt động. Kết quả là dòng kênh canxi giảm đáng kể, tế bào trở nên cạn kiệt canxi và chức năng co bóp sẽ ngừng trong các tế bào đó. Điều này được hy vọng sẽ dẫn đến các bất thường chuyển động thành, đặc biệt ở RVOT, giãn khu vực RVOT và giảm phân suất tống máu ở bệnh nhân có BrS. Nó cũng đã được đề nghị sự mất vòm của điện thế hoạt động, do đó nó tạo ra một trạng thái giống như ngủ đông, có thể, trong một thời gian dài, dẫn đến những thay đổi cấu trúc nhẹ, gồm tích tụ lipid trong tế bào, ngay sát tế bào (vacuolization), cả sự phân phối lại connexin [43]. Những thay đổi về cấu trúc này có thể đóng góp có hiệu quả vào nền gây loạn nhịp của BrS, mặc dù chúng rất khác với những điều được gặp trong bệnh cơ tim thất phải gây loạn nhịp/và loạn sản thất phải gây loạn nhịp (ARVC / D)[31,75]. Giả thuyết này dự đoán một số những thay đổi quan sát được trong những nghiên cứu gần đây có thể là kết quả, chứ không phải nguyên nhân của kiểu hình BrS [76].
Trong nghiên cứu mới gần đây, Nademanee và cộng sự [76] đã thông báo bằng chứng bổ xung tập chung vào các thay đổi bệnh lý trong RVOT của các bệnh nhân BrS đã được chứng minh không thể phát hiện bằng siêu âm tim hoặc MRI.
Ngược lại, các kỹ thuật hình ảnh trong ARVC biểu hiện rõ ràng các thay đổi về hình thái và chức năng (như, giãn, lồi / phình, các bất thường chuyển đồng thành). ARVC là bệnh tim di truyền gây ra thường do proteins desmosom (desmosomal: DS) được phát hiện,[77,78]đặc trưng bằng sự thay thế cơ tim xơ mỡ thúc đẩy rối loạn nhịp thất liên quan đến sẹo có thể dẫn đến SCD, đa số ở người trẻ và vận động viên.[79] Rối loạn nhịp thất nguy hiểm có thể xuất hiện sớm, trong quá trình “pha ẩn” của bệnh, trước khi có thay đổi cấu trúc rõ. [77,78,80]Các nghiên cứu thực nghiệm gần đây cho thấy mất đi biểu hiện của các protein DS có thể gây ra sự bất ổn định điện học tâm thất bằng cách làm rối loạn chức năng kênh natri và sự giảm dòng điện như kết quả của dẫn truyền các tín hiệu không mong muốn của các kênh giữa các phân tử trong các đĩa tiếp nối, làm thúc đẩy loạn nhịp thất nguy hiểm phụ thuộc dòng natri, tương tự đưa đến SCD ở các bệnh nhân hội chứng sóng J. [80-82] Các bằng chứng tiếp theo của sự chồng chéo giữa biểu hiện kiểu hình của ARVC và BrS xuất phát từ (1) các nghiên cứu bệnh học lâm sàng chỉ ra một tập hợp con của các bệnh nhân ARVC có thể và BRS Các bệnh nhân ARVC có thể phân chia các thay đổi ECG và mẫu rối loạn nhịp thất với BrS, [83] và (2) các nghiên cứu tương quan genotype – phenotype chứng minh đột biến PKP2 có thể gây ra phenotype Brugada ở tim người bằng giảm đi dòng natri.[84]Những phát hiện này ủng hộ khái niệm các đột biến gene DS chuyên biệt liên quan đến bệnh học của ARVC có thể đưa đến giới hạn khử cực giảm biểu hiện như sóng J / BrS. Do đó, các hội chứng sóng J và ARVC và J không phải là các trạng thái khác biệt hoàn toàn mà là những điểm kết thúc của một loạt các bất thường cơ tim và thiếu hụt dòng natri có cùng nguồn gốc như các bệnh liên quan đến nhau.[84] Các bất thường về ECG trong ARVC không xác định và biểu hiện sóng T lộn ngược, sóng epsilon và trong giai đoạn tiến triển, giảm biên độ R. Giai đoạn cuối của ARVC thường kết hợp với VT đơn hình với hình thái block nhánh bó trái và được thúc đẩy bằng catecholamine, [85] trong khi BrS được kết hợp với VT đa hình chiếm ưu thế trong quá trình ngủ hoặc lúc nghỉ [86]. Test thử ajmaline dương tính đã được báo cáo ở 16% bệnh nhân ARVC [87,88].
Các yếu tố điều biến
Cân bằng thần kinh, hormone, các yếu tố chuyển hóa và các tác nhân dược lý được cho là điều biến không chỉ hình thái ECG mà còn giải thích sự phát triển rối loạn nhịp thất trong điều kiện nhất định.[89] Bất cứ yếu tố điều biến nào, nếu có, cần được điều chỉnh kịp thời (Bảng 3B).
Mẫu Brugada mắc phải và các phiên bản tương tự
ECG Brugada thường bị che dấu và có thể bị bộc lộ với nhiều loại thuốc và các điều kiện khác nhau, gồm trạng thái sốt, các thuốc hoặc các nghiệm pháp cường phế vị, các kháng aderenergic, chẹn adrenergic, các thuốc chống loạn nhịp Class IC, các thuốc chống trầm cảm tricyclic hoặc tetracyclic, tăng kali máu, hạ kali máu, tăng can xi máu, ngộ độc alcohol và coacine. [90 – 100] Kích thích sớm RV có thể bộc lộ phenotype BrS trong các trường hợp RBBB [101]. Một danh sách cập nhật các thuốc được biết làm bộc lộ ECG Brugada nên được tránh ở các bệnh nhân có BrS có thể tìm trong website www.brugadadrugs.org. [89]
Các yếu tố môi trường dẫn đến sự xuất hiện của một ECG tương tự hoặc giống hệt với mẫu BrS type1 khi không có bất kỳ rối loạn di truyền nào rõ ràng đã được gợi ý để đại diện một phiên bản ECG Brugada.[102] Các đặc tính của các phiên bản Brugada gồm (1) mẫu ECG giống Brugada; (2) sự hiện diện của trạng thái nền có thể khác biệt; (3) sự biến mất của mẫu ECG sau loại bỏ điều kiện; (4) không có tiền sử gia đình có đột tử ở những người thân thế hệ thứ nhất tương đối trẻ (≤ 45 tuổi) hoặc mẫu BrS type 1; (5) không có các triệu chứng như ngất, co giật, hoặc hô hấp ngắc ngoải về ban đêm; và (6) các test thử chẹn kênh natri âm tính. Cuộc tranh luận tiếp tục về sự phù hợp của thuật ngữ này vì rất khó để loại trừ một khuynh hướng di truyền, điều này là một điều kiện tiên quyết để xác nhận biểu hiện ECG như một phiên bản (phenocopy). Việc chỉ định các trạng thái này dưới dạng mẫu ECG Brugada mắc phải hoặc BrS có thể phù hợp hơn và các mô hình ECG Brugada hoặc BrS có thể thích hợp hơn và chính thống hơn với thuật ngữ đã được sử dụng trong hội chứng QT dài.
Cập nhật chẩn đoán ERS
ERS thường được chẩn đoán ở những bệnh nhân có biểu hiện ER ở các chuyển đạo dưới và/hoặc bên biểu hiện với ngừng tim được cứu sống, VF, hoặc VT đa hình được chứng minh bằng tư liệu. Phù hợp thông báo đồng thuận mới đây về ERP, [24] ER được ghi nhận nếu (1) có kết thúc của QRS chẽ đôi (sóng J) hoặc líu díu ở chỗ xuống của sóng R chiếm ưu thế có hoặc không có sự chênh lên của đoạn ST; (2) đỉnh chẽ đôi hoặc sóng J (Jp) ≥0,1 mV ở ≥2 chuyển đạo liên tiếp của ECG 12 chuyển đạo, không bao gồm các chuyển đạo V1 -V3; và (3) khoảng thời gian QRS (được đo ở các chuyến đạo ở đó chẽ đôi hoặc líu díu không có) <120 ms. Bảng 4 liệt kê các tiêu chí loại trừ trong chẩn đoán phân biệt của ERS.
Hệ thống điểm số chẩn đoán được đề nghị cho ERS, được gọi là Thang điểm ERS Thượng Hải, được trình bày trong Bảng 5. Hệ thống tính điểm dựa trên bằng chứng có sẵn trong tư liệu cho đến nay. Như trong BrS, cân nhắc các biến số dựa trên ý kiến chuyên gia được thông báo từ các nghiên cứu thuần tập mà không bao gồm tất cả các biến được trình bày. Do đó, các hệ số được cân nhắc nghiêm ngặt, khách quan không có nguồn gốc từ các tập dữ liệu về các yếu tố nguy cơ và kết quả. Tuy nhiên, các tác giả tin rằng một thông số cân nhắc suy diễn có thể có lợi khi áp dụng cho bệnh nhân. Như với tất cả các khuyến cáo như vậy, họ sẽ cần phải trải qua quá trình xác nhận ban đầu và đang diễn ra trong các nghiên cứu trong tương lai.
Những điểm tương đồng và sự khác biệt giữa BrS và ERS
BrS và ERS hiển thị một số điểm tương đồng về lâm sàng, gọi ý sinh bệnh lý tương tự (Bảng 6) [19,21,103- 105] Nam giới chiếm ưu thế trong cả hai hội chứng, với BrS chiếm 71% – 80% ở người da trắng và 94% -96% ở người Nhật. [106,107] Trong trạng thái ERP, VF xảy ra chủ yếu ở nam giới (72%) khi nghiên cứu trong một nghiên cứu thuần tập quốc tế [2] nhưng ở một tỷ lệ phần trăm cao hơn trong một báo cáo của các nhà khám phá Nhật Bản [108] Bệnh nhân BrS và ERS có thể hoàn toàn không có triệu chứng cho đến khi họ bị ngừng tim. Trong cả hai hội chứng, tần suất xuất hiện VF hoặc SCD cao nhất xảy ra trong thập niên thứ ba của đời sống, có lẽ liên quan đến nồng độ testosterone ở nam giới [109]. Trong cả hai hội chứng, sự xuất hiện của các sóng J và chênh lên của đoạn ST thường kết hợp với nhịp chậm hoặc khoảng ngừng [110,111] Điều này có thể giải thích tại sao VF ở cả hai hội chứng thường xảy ra trong khi ngủ hoặc trong một số hoạt động thể lực mức độ thấp [108,112] Khoảng QT tương đối ngắn ở bệnh nhân ERS, [2,113] và BrS có mang đột biến ở gen kênh canxi.[114]
Như sẽ được thảo luận chi tiết hơn sau này, ERS và BrS cũng có những điểm tương đồng về đáp ứng với điều trị thuốc. Trong cả hai, các cơn bão điện và các biểu hiện sóng J liên quan có thể được ngăn chặn bằng các chất chủ vận b- adrenergic. [115 – 118] Phác đồ điều trị bằng thuốc uống quinidine, [119.120] bepridil, [117] denopamine, [115,121] và cilostazol lâu dài [115,117,121-125] để ngăn chặn phát triển của VT / VF trong cả ERS và BrS phụ thuộc vào ức chế Ito, gia tăng Ica, hoặc cả hai. [3,122,126]
Sự khác nhau giữa 2 hội chứng bao gồm (1) khu vực tim bị ảnh hưởng nhất (RVOT đối lại LV thấp hơn); (2) sự hiện diện của các bất thường cấu trúc (rời rạc) trong BrS nhưng không có trong ERS; (3) tần suất điện thế trễ trong ECGs tín hiệu trung bình (BrS 60% [4] ERS 7%) [108]; và (4) sự chênh lên của Jo, Jp, Jt lớn hơn (chênh lên của đoạn ST) trong đáp ứng với các thuốc chẹn kênh natri trong BrS và ERS và tỷ lệ rung nhĩ (AF) cao hơn trong BrS đối lại với ERS.[127] Các nghiên cứu ban đầu đã gợi ý nền tảng sinh lý bệnh khác biệt cho ERS và BrS dựa trên cơ sở quan sát các blockers kênh natri làm bộc lộ hoặc làm nổi bật biểu hiện sóng J trong BrS nhưng làm giảm biên độ trong ERS [108]. Tuy nhiên, nghiên cứu gần đây của Nakagawa và cộng sự [357] cho thấy các sóng J được ghi lại bằng cách sử dụng các điện cực đơn cực thượng tâm mạc hướng vào tĩnh mạch vành bên trái ở các bệnh nhân ERS đã thực sự tăng lên, ngay cả khi các sóng J được ghi ở các chuyển đạo trước tim bên bị giảm xuống, chủ yếu do sự hấp thụ sóng J bề mặt bằng QRS mở rộng [29.108] Báo cáo trường hợp của Nakagawa và cộng sự gần đây đã được bổ sung thêm các trường hợp trong đó kỹ thuật này đã được sử dụng; 2 trong số 3 trường hợp này chỉ ra sự nổi bật của các sóng J được tạo ra do pilsicainide trên điện tâm đồ được ghi từ bề mặt thượng tâm mạc của LV (H. Morita, những quan sát chưa được công bố). Ngoài ra, để hỗ trợ cho luận đề này các mô hình và mẫu ECG và hội chứng này liên quan chặt chẽ đến các báo cáo về các trường hợp ERS chuyển tiếp vào ERS và BrS. [105,128]
Sự khác biệt cơ bản giữa BrS và ERS có liên quan đến vùng tâm thất bị ảnh hưởng nhiều nhất. Các nghiên cứu lập bản đồ thượng tâm mạc ở bệnh nhân BrS báo cáo các sóng J được nổi lên và được phân đoạn và / hoặc điện thế trễ ở khu vực thượng tâm mạc của RVOT, [129 – 131] trong khi ở ERS chỉ có sóng J nổi lên, đặc biệt ở thành dưới LV. [29] Hoạt động điện đồ được phân chia và điện thế trễ đã được quan sát trên mô hình thực nghiệm của ERS [30] nhưng vẫn chưa được báo cáo về mặt lâm sàng. Các nghiên cứu điện giải phẫu lập bản đồ không xâm lấm đã báo cáo độ chênh tái cực được khu trú rất dốc đứng qua các khu vực dưới / bên của LV ở các bệnh nhân ERS, được đi trước bằng hoạt động thất bình thường,[132] trong khi ở BrS cả hai dẫn truyền không liên tục và sự phân tán dốc đứng của tái cực được thể hiện ở RVOT.[133] Một khác biệt được giả định khác là sự hiện diện của các bất thường cấu trúc trong BrS, nhưng còn không được mô tả trong ERS [76].
Mặc dù sóng J được lộ ra hoặc được tạo ra bằng cả hai hạ thân nhiệt và sốt, sự phát triển rối loạn nhịp trong ERS nhạy cảm nhiều hơn với hạ thân nhiệt, còn sự rối loạn nhịp ở BrS dường như chỉ được thúc đẩy bằng sốt. [33,34,134,135,140] Hạ thân nhiệt đã được thông báo làm tăng nguy cơ VF trong ERS, [33.34.134.135.140] và được ghi nhận rõ như là một yếu tố nguy cơ lớn trong BrS.[138,139] Cần lưu ý hạ thân nhiệt có thể làm giảm biểu hiện của ECG của BrS khi đã có biểu hiện.[141,142]
ERP có liên quan với nguy cơ tăng VF ở bệnh nhân nhồi máu cơ tim cấp tính [143]và hạ thân nhiệt. [33,144] ERP tự nhiên ở các chuyển đạo bên-dưới cũng đã được thông báo được kết hợp với nguy cơ tăng lên của các biến cố loạn nhịp ở các bệnh nhân BrS. Kawata và cộng sự[145]đã thông báo tỷ lệ ER ở các chuyển đạo bên – dưới cao (63%) ở bệnh nhân BrS bị VF được chứng minh bằng tư liệu.
Bảng 4 Chẩn đoán phân biệt của mẫu tái cực sớm
|
Các nguyên nhân khác của mẫu tái cực sớm gồm các nguyên nhân sau: |
|
† Mẫu ST ở vị thành niên † Bệnh màng ngoài tim (viêm màng ngoài tim, kén màng ngoài tim, u màng ngoài tim) † Hạ thân nhiệt † Tăng thân nhiệt † U cơ tim (lipoma) † Bệnh tim do tăng huyết áp † Tim của vận động viên † Thiếu máu cơ tim cục bộ † Nhồi máu cơ tim ST chênh lên (STEMI) (như nhồi máu cơ tim trước vách) † QRS phân đoạn (chẽ đôi phần cuối) † Giảm can xi máu † Tăng kali máu † U tuyến ức (Thymoma) † Bóc tách động mạch chủ † Bệnh cơ tim thất phải gây rối loạn nhịp † Bệnh cơ tim Takotsubo † Các nguyên nhân thần kinh (chảy máu nội sọ, tổn thương não cấp) † Viêm cơ tim † Bệnh Chagas † Sử dụng Cocaine |
Bảng 5: Hệ thống thang điểm Thượng Hải được đề xuất cho chẩn đoán hội chứng tái cực sớm
|
|
Điểm |
|
I. Bệnh sử lâm sàng |
|
|
A. Ngừng tim không thể giải thích, VT hoặc VT đa hình được chứng minh bằng tư liệu |
3 |
|
B. Ngất được nghi ngờ do rối loạn nhịp |
2 |
|
C. Ngất cơ chế chưa rõ / bệnh căn không rõ |
1 |
|
*Chỉ thưởng điểm một lần cho điểm cao nhất trong phạm vi danh mục này |
|
|
II. ECG 12 chuyển đạo |
|
|
A. ER ≥0.2 mV ở ≥2 chuyển đạo dưới và hoặc bên với đoạn ST đi ngang /đi xuống |
2 |
|
B. Các thay đổi động học ở sự chênh lên của điểm J (≥0,1 mV) ở ≥2 chuyển đạo dưới và /bên của ECG |
1,5 |
|
C. Chênh lên của điểm J ≥ 0,1 mV ở ít nhất 2 chuyển đạo dưới và /hoặc bên cảu ECG |
1 |
|
*Chỉ thưởng điểm một lần cho điểm cao nhất trong phạm vi danh mục này |
|
|
III. Theo dõi ECG lưu động |
|
|
A. PVCs khoảng ghép ngắn với R lên trên rìa đi lên hoặc đỉnh sóng T |
2 |
|
IV. Bệnh sử gia đình |
|
|
A. Người thân với ERS xác định |
2 |
|
B. ≥2 người thân thế hệ thứ nhất với mẫu ECG II.A. |
2 |
|
C. Người thân thế hệ thứ nhất với mẫu ECG II.A. |
1 |
|
D. Đột tử tim < 45 tuổi không thể giải thích ở người thân thế hệ thứ nhất và thứ hai |
0,5 |
|
*Chỉ cộng thêm điểm một lần cho thang điểm cao nhất trong mục này |
|
|
V. Kết quả test di truyền |
|
|
A. Đột biến mẫn cảm có khả năng gây bệnh ERS |
0,5 |
|
Thang điểm (Đòi hỏi ít nhất 1 dấu hiệu ECG) |
|
|
≥ 5 điểm: Có khả năng / ERS xác định |
|
|
3–4.5 điểm: ERS có thể |
|
|
< 3 điểm: Không được chẩn đoán |
|
ER = tái cực sớm; ERS = Hội chứng tái cực sớm; PVC = co bóp thất sớm; VF = rung thất; VT = nhịp nhanh thất.
Bảng 6. Những điểm tương đồng và khác biệt giữa hội chứng Brugada và hội chứng tái cực sớm và các cơ chế cơ bản có thể xảy ra
|
|
BrS |
ERS |
Cơ chế có thể(s) |
|
Các tương đồng giữa BrS và ERS |
|
|
|
|
Chiếm ưu thế ở nam |
Có (475%) |
Có (480%) |
Testosterone điều biến các dòng ion làm nền cho sự chẽ đôi của AF thượng tâm mạc |
|
Tuổi trung bình của biến cố đầu tiên |
30–50 |
30–50 |
|
|
Kết hợp với các đột biến hoặc các biến thể hiếm trong KCNJ8, CACNA1C, CACNB2, CACNA2D, SCN5A, ABCC9, SCN10A |
Có |
Có |
Gia tăng chức năng các dòng đi vào (IK-ATP) hoặc mất chức năng các dòng đi vào chảy vào trong (ICa hoặc INa) |
|
Các khoảng QT tương đối ngắn ở những người có đột biến kênh Ca |
Có |
Có |
Mất chức năng ICa |
|
Động học ECG |
Cao |
Cao |
Điều biến tự động các dòng kênh ion là nền các pha sớm của AF thượng tâm mạc |
|
VF thường xuất hiện trong quá trình ngủ hoặc ở mức hoạt động thể lực thấp |
Có |
Có |
Mức trương lực phế vị cao hơn của Ito ở tần số tim chậm hơn |
|
Khởi kích VT/VF |
PVC khoảng ghép ngắn |
PVC khoảng ghép ngắn |
Vào lại Pha 2 |
|
Đáp ứng cải thiện với quinidine và bepridil |
Có |
Có |
Ức chế Ito và có thể hiệu quả tiêu phế vị |
|
Đáp ức cải thiên với isoproterenol denopamine và milrinone |
Có |
Có |
ICa được tăng lên và tần số tim nhanh hơn |
|
Đáp ứng cải thiện với cilostazol |
Có |
Có |
ICa được tăng lên, Ito bị giảm xuống và tần số tim nhanh hơn |
|
Đáp ứng cải thiện với tạo nhịp |
Có |
Có |
Khả năng Ito bị giảm do làm chậm phục hồi sự không hoạt hóa |
|
Bộ lộ mẫu ECG qua trung gian phế vị |
Có |
Có |
Hiệu quả trực tiếp làm ức chế ICa và gián tiếp làm tăng Ito (do làm chậm tần số tim) |
|
Ảnh hưởng của các chẹn kênh natri trên điện đồ thượng tâm mạc đơn cực |
Các sóng J được tăng thêm |
Các sóng J được tăng thêm |
Sự chuyển ra bên ngoài của cân bằng dòng trong giai đoạn đầu của AP thượng tâm mạc |
|
Sốt |
Các sóng J được tăng thêm |
Các sóng J được tăng thêm (hiếm) |
Không có hoạt động INa được gia tăng và phục hồi Ito từ không hoạt động được gia tăng |
|
Hạ thân nhiệt |
Các sóng J gia tăng bắt chước Brs |
Các sóng J gia tăng |
Hoạt động chậm của ICa, để lại Ito không bị cản trở. Vào lại pha 2 tăng lên nhưng pVT bị giảm do kéo dài APD [358] |
|
Sự khác biệt giữa BrS và ERS |
|
|
|
|
Khu vực được liên quan nhất |
RVOT |
Thành dưới LV |
Các mức độ của Ito và / hoặc sự khác biệt trong dẫn truyền cao hơn |
|
Các chuyển đạo bị ảnh hưởng |
V1 –V3 |
II, II a, VF, V4, V5, V6; I, aVL, Cả hai: bên-dưới |
|
|
Sự khác biệt trong tỷ lệ khu vực |
|
|
Châu Âu: BrS = ERS Châu Á: BrS 4 ERS |
|
Tần suất của điện thế trễ trong ECG tin hiệu trung bình |
Cao hơn |
Thấp hơn |
|
|
Tỷ lệ rung nhĩ |
Cao hơn |
Thấp hơn |
|
|
Hiệu quả của block kênh natri trên ECG bề mặt |
Biểu hiện sóng J được tăng lên |
Biểu hiện sóng J bị giảm đi |
Giảm sóng J trong trạng thái ER được cho là do phần lớn là kéo dài QRS. Sự nổi lên của các khiếm khuyết tái cực chiếm ưu thế trong BrS, trong khi đó sự nổi lên của các khiếm khuyết khử cực lại chiếm ưu thế hơn trong ERS. |
|
Những thay đổi về cấu trúc, bao gồm xơ hóa nhẹ và giảm biểu hiện của Cx43 trong sự thâm nhiễm RVOT hoặc thâm nhiễm xơ mỡ trong các trường hợp bệnh cơ tim thất phải gây loạn nhịp. Các nghiên cứu hình ảnh học cũng cho thấy những bất thường về chuyển động thành và sự giãn nở nhẹ ở khu vực RVOT. |
Cao hơn trong một số hình thái của hội chứng |
Chưa biết |
Một số nhà nghiên cứu đã đưa ra giả thuyết một số thay đổi này có thể là kết quả của chứ không phải là nguyên nhân của chất nền BrS, có thể tạo ra một trạng thái giống ngủ đông do mất sự co bóp trong RVOT thứ phát do mất vòm AP. |
AP = điện thế hoạt động; APD = khoảng thời gian điện thế hoạt động; BrS = hội chứng Brugada; ERS = hội chứng tái cực sớm; RVOT = đường ra thất phải; PVC = co bóp thất sớm; pVT = nhanh thất đa hình; VF = rung thất; VT = nhanh thất.
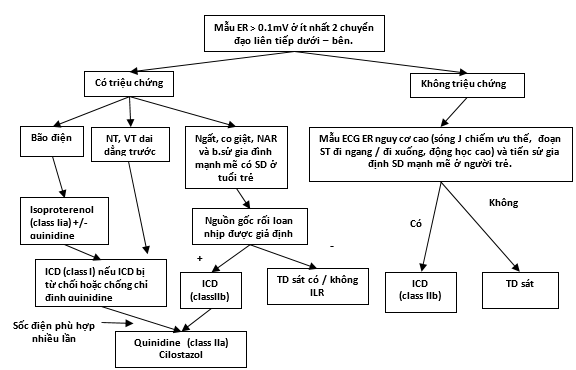
Hình 7. Chỉ định điều trị các bệnh nhân có hội chứng tái cực sớm. Các khuyến cáo với chỉ dẫn Class nhận được từ Priori SG, Wilde AA, Horie M, và cộng sự. Báo cáo đồng thuận của chuyên gia HRS / EHRA / APHRS về việc chẩn đoán và điều chỉnh các bệnh nhân có hội chứng rối loạn nhịp di truyền tiên phát: các tài liệu đã được HRS, EHRA và APHRS xác nhận vào tháng 5 năm 2013 và ACCF, AHA, PACES, AEPC vào tháng 6 năm 2013. Nhịp Tim năm 2013, 10: 1932-1963, và Priori SG, Blomstrom-Lundqvist C, Mazzanti A, và cộng sự. Hướng dẫn của ESC năm 2015 về điều chỉnh các bệnh nhân rối loạn nhịp thất và ngăn ngừa đột tử tim: Nhóm Công tác cho Điều chỉnh Các Bệnh nhân rối loạn nhịp thất và Ngăn ngừa Đột tử Tim của Hội Tim Mạch châu Âu (ESC). Đã được Hội Tim Mạch Bẩm sinh và Nhi khoa châu Âu (AEPC) xác nhận. Tạp chí Tim châu Âu 2015;36: 2757–9. Các khuyến cáo không chỉ dẫn Class được đưa ra từ sự đồng thuận của các tác giả.. ER = tái cực sớm; ICD = máy khử rung tim có thể cấy; ILR = Ghi vi mạch có thể cấy; NAR = hô hấp ngắc ngoải về ban đếm; VT = nhịp nhanh thất. NT: ngưng tim. TD: theo dõi.
Máy khử rung tim có thể cấy
Chiến lược điều trị hiệu quả duy nhất đã được chứng minh để phòng ngừa SCD ở bệnh nhân BrS và ERS có nguy cơ cao là ICD. [301,302] Điều quan trọng phải nhận ra ICDs có liên quan đến các biến chứng, đặc biệt ở những người trẻ hoạt động. [249,303] Trong 10 năm sau khi cấy ghép, tỷ lệ sốc không phù hợp và đưa đến thất bại lần lượt là 37% và 29%. Theo dõi từ xa có thể xác định hướng đến thất bại và ngăn ngừa các cú sốc không thích hợp [304]. ICDs dưới da được cho là đại diện cho tương lai của chỉ định này do chúng được cho liên quan ít đến biến chứng trong cuộc sống [305]
Cấy ICD là điều trị hàng đầu cho các bệnh nhân JWS biểu hiện SCD được cứu sống hoặc VT/VF được chứng minh bằng tư liệu có hoặc không có ngất (khuyến cáo Class I). [301,306] ICD có thể hữu ích (Class IIa) ở bệnh nhân có BrS có triệu chứng với mẫu type 1, ở người ngất nhiều khả năng do VT/VF. Các chuyên gia HRS / EHRA / APHRS đồng ý ICD có thể được xem xét (Class IIb) ở những bệnh nhân không triệu chứng với VF có thể tạo ra trong quá trình kích thích điện có chương trình (PES). [8] Một số nghiên cứu gợi ý giá trị tiên đoán của các nghiên cứu EP có thể được cải thiện bằng cách giới hạn quy trình PES tới 2 kích thích ngoài [50,276] nhưng quan sát này không được hỗ trợ bằng các nghiên cứu khác. [49,307] Tương tự, một số nghiên cứu cho PES nên được giới hạn ở RVA và cho rằng chiến lược PES bị hạn chế này cho giá trị tiên đoán dương cao được tìm thấy ở một vài loạt. [275] Một lần nữa, quan sát này lại không được xác nhận bằng các nghiên cứu khác [276].
Nhóm Công Tác hiện tại đề xuất ICDs hợp lý (Class IIa) ở bệnh nhân BrS có triệu chứng với mẫu type 1 nhưng việc cấy được xem xét theo từng trường hợp do một bác sỹ điện sinh kinh nghiệm trong BrS, có tính đến tuổi, giới tính, thể hiện lâm sàng, các đặc điểm ECG (QRS phân đoạn, biên độ Jp), và sở thích của bệnh nhân. Nhóm Công Tác hiện tại cũng đề nghị nghiên cứu EP có thể được xem xét ở các cá thể không triệu chứng với mẫu type 1 Brugada tự phát. Nếu VT / VF có thể tạo ra được, cần xem xét một ICD. [7] Các nghiên cứu gần đây cho thấy sử dụng ≤ 2 kích thích ngoài để gây VT / VF. [50,276] Các ICDs không được chỉ định ở những bệnh nhân không có triệu chứng mà không có bất kỳ đặc điểm này. Hiện tại, không có vai trò rõ ràng đối với PES ở bệnh nhân ERS.
Điều trị máy tạo nhịp
Các biến cố rối loạn nhịp và SCD ở cả hai BrS và ERS thường biểu hiện lúc ngủ hoặc khi nghỉ và kết hợp với nhịp tim chậm. Các quan sát này không đứng vững, vai trò tiềm tàng cho tạo nhịp tim còn chưa có đột phá lớn. [308] Có rất ít ca thông báo có sẵn.[309,310]
Điều trị RFA
Nademanee và cộng sự [129] cho thấy RFA các vị trí thượng tâm mạc có biểu hiện điện thế trễ và điện đồ lưỡng cực phân đoạn ở RVOT ở bệnh nhân BrS có thể làm giảm đáng kể tình trạng loạn nhịp và biểu hiện ECG của bệnh. Việc triệt phá ở các vị trí này đã được thông báo tại các địa điểm này đã làm cho VT / VF không thể tạo ra và làm bình thường mẫu ECG BrS ở đa số bệnh nhân trong một tuần hoặc vài tháng. Theo dõi dài hạn (20-6 tháng) không có VT / VF tái phát, chỉ có 1 bệnh nhân điều trị bằng amiodarone. Báo cáo trường hợp ủng hộ những hiệu quả này đã được xuất bản. [311] Các bằng chứng bổ sung để hỗ trợ hiệu quả của việc triệt phá nền thượng tâm mạc được Sacher và cộng sự [130], Shah và cộng sự [312] cung cấp.
Mới đây, Brugada và cộng sự [131] đã sử dụng flecainide để xác định toàn bộ phạm vi hoạt động điện đồ điện thế thấp ở trước RV và RVOT cũng như nhằm vào khu vực này cho RFA. Trong tất cả 14 bệnh nhân BRS, RFA loại bỏ các điện đồ lưỡng cực bất thường, chênh lên của đoạn ST được bình thường hóa trên các chuyển đạo trước tim phải của ECG, và VT / VF không còn có thể tạo ra được nữa. Liệu pháp triệt phá có thể được xem xét (khuyến cáo Class IIb) ở các bệnh nhân BrS với sốc ICD phù hợp thường xuyên do bão điện tái phát. [7] Không có báo cáo lâm sàng về việc triệt phá nền LV ở bệnh nhân ERS. Ở những bệnh nhân BrS kết hợp với ERS, việc triệt phá thượng tâm mạc RV trước (gồm RVOT) không hiệu quả.
Tiếp cận dược lý cho điều trị
Hội chứng Brugada
Cấy ICD có thể là vấn đề ở trẻ sơ sinh hoặc trẻ nhỏ do tần số biến chứng cao. Các ICDs cũng ngoài khả năng kinh tế để đạt được ở một số bệnh nhân ở một số khu vực trên thế giới. Cách tiếp cận thuốc đối với liệu pháp, dựa trên sự tái cân bằng dòng hoạt động trong giai đoạn đầu của AP thượng tâm mạc ở RV để giảm độ lớn chẽ đôi của AP và / khôi phục vòm AP, đã là trọng tâm của nghiên cứu cơ bản và lâm sàng trong những năm gần đây. Các thuốc chống loạn nhịp chẳng hạn như amiodarone và beta-blockers đã tỏ ra không có hiệu quả [313]. Các thuốc chống loạn nhịp loại IC (ví dụ: flecainide, propafenone) và các tác nhân IA (ví dụ như procainamid) đều bị chống chỉ định vì những ảnh hưởng của chúng làm bộc lộ BrS gây ra và gây ra rối loạn nhịp tim. Disopyramide là thuốc chống loạn nhịp class IA đã được chứng minh làm bình thường đoạn ST chênh lên ở một số bệnh nhân BrS nhưng không làm bộc lộ hội chứng ở những bệnh nhân khác [314]
Do sự hiện diện của Ito chiếm ưu thế là điều kiện tiên quyết cho sự phát triển cả BrS và ERS, việc ức chế một phần dòng này được cho là có hiệu quả bất kể nền tảng ion hay gen cho bệnh. Thật không may, các thuốc blocker chọn lọc lên tim và blocker chuyên biệt Ito không có hiệu lực.
Chỉ thuốc với các đặc tính block Ito có ý nghĩa hiệu lực ở Hoa Kỳ và trên thế giới là quinidine. [19,73] Các nghiên cứu thực nghiệm đã chỉ ra quinidine có hiệu quả trong việc khôi phục lại vòm AP trên cơ tim, do đó bình thường hóa đoạn ST và ngăn ngừa vào lại pha 2 và VT đa hình trong một loạt các mô hình thí nghiệm khác nhau của BrS. [19,150,315 – 317] Một nghiên cứu thực nghiệm gần đây cho thấy quinidine, do hiệu quả block Ito, cũng có thể sử dụng hiệu quả bảo về chống lại VT/VF thúc đẩy do hạ thân nhiệt trong mô hình JWS [144]. Đáng chú ý, theo lịch sử, quinidine được dùng để ngăn ngừa VF ở những bệnh nhân cần hạ nhiệt cho các thủ thuật ngoại khoa. [317]
Bằng chứng lâm sàng cho thấy hiệu quả của quinidine trong việc bình thường sự chênh lên của đoạn ST và / hoặc ngăn ngừa các biến cố loạn nhịp ở bệnh nhân BrS đã được báo cáo trong nhiều nghiên cứu và các báo cáo trường hợp. [117,119,120,124,318 – 331] Hermida và cộng sự [119] báo cáo hiệu quả 76% trong phòng ngừa VF do PES gây ra. Belhassen và cộng sự [332] gần đây đã báo cáo hiệu quả 90% trong việc ngăn ngừa khởi phát VF sau khi điều trị bằng quinidine mặc dù đã sử dụng các phương pháp kích thích rất mạnh mẽ. Hơn nữa, không có sự cố loạn nhịp xảy ra giữa các bệnh nhân BrS được điều trị bằng quinidine trong thời gian theo dõi trung bình 10 năm.
Trong một thử nghiệm gần đây được thực hiện tại 2 trung tâm ở Pháp, 44 bệnh nhân BrS không triệu chứng với có thể thúc đẩy VT / VF đã được ghi nhận (47-10 tuổi, 95% nam) .[333] Trong số những bệnh nhân này, 34 (77%) không còn thúc đẩy được nữa trong khi điều trị bằng hydroquinidine 600 mg / ngày trong 6,2-3 năm. Trong số 10 bệnh nhân khác (22%) vẫn còn bị thúc đẩy và đã nhận ICD (nhóm PVS +), không ai được điều trị thích hợp trong thời gian theo dõi trung bình 7,7-2 năm.
Một đăng ký tiềm cứu của quinidine theo kinh nghiệm đối với BrS không triệu chứng đã được tính toán. Nghiên cứu này được đăng tải trên trang web của Cơ quan Y tế Quốc gia (ClinicalTrials.gov) và có thể truy cập tại http://clinicaltrials.gov/ct2 /show/NCT00789165?term_brugada&rank_2. Liều từ 600 đến 900 mg đã được khuyến cáo, nếu được dung nạp [322].
Quinidin có thể được xem xét (chỉ định class IIb) ở bệnh nhân BrS có các cơn bão điện và ở những bệnh nhân đã được cấy ICD đang có những cú sốc thích hợp lặp lại. Quinidin cũng có thể hữu ích ở các bệnh nhân BrS không có triệu chứng biểu hiện ECG type 1 tự phát, nếu họ hạn chế cho ICD và thiết bị này bị từ chối hoặc chống chỉ định (khuyến cáo của Class IIa).
Các thuốc làm gia tăng dòng kênh canxi typ L, như các chất beta-adrenergic (ví dụ isoproterenol, denopamine, orciprenaline) cũng hữu ích. [19,117,121,327,334,335] Isoproterenol đôi khi kết hợp với quinidine đã được sử dụng thành công để kiểm soát bão VF và bình thường hóa sự chênh lên ST, đặc biệt ở trẻ em. [93,115 – 118,279,319,320,325,331,336 – 343] VF tự phát ở bệnh nhân BrS thường liên quan đến tăng trương lực phế vị và phù hợp với điều trị bằng sự gia tăng trương lực giao cảm qua việc sử dụng isopro-przestrzol. Việc sử dụng isoproterenol là một khuyến cáo Class IIa đối với bệnh nhân BrS có các cơn bão điện [7].
Một phương pháp tiếp cận dược lý đầy hứa hẹn khác đối với BrS là sử dụng thuốc cilostazol ức chế phosphodiesterase III [117,121,123] làm bình thường hóa đoạn ST, có thể do tăng thêm dòng canxi (ICa) cũng như bằng cách giảm Ito thứ phát đưa đến tăng cAMP và nhịp tim. [344] Các ảnh hưởng khác của cilostazol có thể góp phần vào các hoạt động của nó (ví dụ: adenosine, NO, IKATP ty thể [345]). Hiệu quả của nó kết hợp với bepridil trong việc ngăn ngừa các đợt VF đã được Shimohara và cộng sự báo cáo gần đây. [125] Sự thất bại của cilostazol trong điều trị BrS đã được mô tả trong một báo cáo trường hợp đơn lẻ [346]. Milrinone là một chất ức chế phosphodiesterase III gần đây được xác định là một thay thế mạnh hơn cho cilostazol trong việc ức chế sự chênh lên của ST và loạn nhịp trong mô hình thí nghiệm BrS. [150,347] Còn chưa có báo cáo nào về lâm sàng.
Wenxin Keli, một thuốc cổ truyền của Trung Quốc, mới đây đã chỉ ra ức ức Ito và do đó ức chế VT đa hình trong mô hình thực nghiệm BrS khi phối hợp với nồng độ thấp quinidine (5 mM).[316]
Các chất làm tăng đỉnh và INa muộn, bao gồm bepridil và dimethyl lithospermate B, được gợi ý có giá trị trong BrS. Bepridil đã được báo cáo để ngăn chặn VT / VF trong một số nghiên cứu ở bệnh nhân BrS. [117,297,298,348] Hoạt động của thuốc được cho là trung gian bằng (1) ức chế Ito; (2) sự gia tăng INa thông qua việc điều chỉnh các kênh natri [349]; và (3) kéo dài khoảng QT khi tần số chậm do tăng độ dốc QT/RR. [297,298] Dimethyl lithospermate B, một chất chiết xuất của Danshen, phương thuốc thảo dược truyền thống Trung Quốc, đã được báo cáo làm chậm bất hoạt của INa, do đó tăng INa trong suốt các giai đoạn đầu của AP và ngăn chặn loạn nhịp trong mô hình thí nghiệm của BrS [350].
Do các rối loạn nhịp thất ác tính ít gặp ở các bệnh nhân BrS không triệu chứng [247] hoặc ERP[44] và thường không liên quan với hoạt động thể lực, sự hiện diện của các mẫu này không phải chống chỉ định tham gia thi dấu thể thao, mặc dù, như đã thảo luận trước, hiện nay tư liệu không đầy đủ để có thể làm các khuyến cáo xác định cho tham gia thể thao.
Hội chứng tái cực sớm
Không có gì đáng ngạc nhiên khi phương pháp tiếp cận điều trị ERS tương tự như của BrS, bởi vì các cơ chế nền tảng 2 hội chứng có thể tương tự nhau. Quinidin, chất ức chế phosphodiesterase III, và isoproterenol đã được chứng minh là có tác dụng cải thiện trong việc ngăn ngừa hoặc làm mất hoạt động rối loạn nhịp liên quan đến ERS. Isoproterenol đã được chứng minh có hiệu quả trong việc làm ồn các cơn bão điện phát triển ở bệnh nhân BrS [117,338] hoặc ERS [190]. Isoproterenol đã được chứng minh là hoạt động bằng cách đảo ngược các bất thường tái cực do kiểu hình bệnh thứ phát gây ra các kiểu hình bệnh tật thứ phát trong việc phục hồi vòm AP trên cơ sở các mô hình thí nghiệm của BrS [19,315] và ERS [30]. Hoạt động này của chất chủ vận beta-adrenergic được cho do các hoạt động của nó làm tăng mạnh ICa.
Thuốc cilostazol ức chế phosphodiesterase III đã được báo cáo để làm giảm ECG và biểu hiện loạn nhịp của ERS [122]. Các ức chế Phosphodiesterase được biết để hoạt hóa Ica thứ phát do tăng cAMP. [121,344,351 – 355] Sự gia tăng của ICa được cho là ngăn ngừa rối loạn nhịp liên quan đến JWS bằng cách đảo ngược các khiếm khuyết tái cực và khôi phục tính đồng nhất về điện ngang qua vách thất thứ phát để hồi phục vòm AF thượng tâm mạc ở cả hai BrS [347] và ERS. [144] Cilostazol đã được giả thuyết cũng gây block Ito. Sự gia tăng ICa cùng với việc ức chế Ito được mong đợi để tạo ra sự dịch chuyển về phía trong trong sự cân bằng các dòng hoạt động trong giai đoạn đầu của AP thượng tâm mạc, đặc biệt hiệu quả trong việc ức chế hoạt động sóng J. Hiệu quả của bepridil trong ERS đã được báo cáo ở một bệnh nhân duy nhất cho đến nay. [356]
Không có dữ liệu lâm sàng về hiệu quả của RFA trong trạng thái ERS, mặc dù thực tế hoạt động điện đồ điện thế được phân đoạn và điện thế trễ có tần số cao được quan sát trong thất trái ở các bệnh nhân ERS [357] và trong các mô hình thực nghiệm của ERS (Yoon và Antzelevitch, dữ liệu chưa được công bố). Nakagawa và cộng sự [357] đã báo cáo kết quả nghiên cứu trong đó họ ghi lại các điện đồ thượng tâm mạc trực tiếp từ LV của các bệnh nhân được chẩn đoán ERS bằng catheter đa điện cực được hướng dẫn vào tĩnh mạch vành bên (marginal) trái, tĩnh mạch vành liên thất trước, cũng như tĩnh mạch trung tâm tim qua xoang vành. Các tác giả đã báo cáo các điện thế trễ ở các điện đồ lưỡng cực được ghi từ thượng tâm mạc LV của các bệnh nhân ERS [357].
Tài liệu tham khảo
1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol 1992;20:1391–1396.
2. Haissaguerre M, Derval N, Sacher F et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016–2023.
3. Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med 2008;358:2078 2079.
4. Rosso R, Kogan E, Belhassen B, Rozovski U, Scheinman MM, Zeltser D, Halkin A, Steinvil A, Heller K, Glikson M, Katz A, Viskin S. J-point elevation in survivors of primary ventricular fibrillation and matched control subjects: incidence and clinical significance. J Am Coll Cardiol 2008;52:1231–1238.
5. Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002; 106:2514–2519.
6. Antzelevitch C, Brugada P, Borggrefe M et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005;111:659–670.
7. Priori SG, Wilde AA, Horie M et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;15:1389–1406.
8. Priori SG, Wilde AA, Horie M et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May
2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013; 10:1932–1963.
9. Priori SG, Blomstrom-Lundqvist C, Mazzanti A et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2757–2759.
10. Clements SD, Hurst JW. Diagnostic value of ECG abnormalities observed in subjects accidentally exposed to cold. Am J Cardiol 1972;29:729–734.
11. Thompson R, Rich J, Chmelik F, Nelson WL. Evolutionary changes in the electrocardiogram of severe progressive hypothermia. J Electrocardiol 1977;10:67–70.
12. Eagle K. Images in clinical medicine. Osborn waves of hypothermia. N Engl J Med 1994;10:680.
13. Kraus F. Ueber die wirkung des kalziums auf den kreislauf 1. Dtsch MedWochenschr 1920;46:201–203.
14. Sridharan MR, Horan LG. Electrocardiographic J wave of hypercalcemia. Am J Cardiol 1984;54:672–673.
15. Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm 2010;7:549–558.
16. Wasserburger RH, Alt WJ. The normal RS-T segment elevation variant. Am J Cardiol 1961;8:184–192.
17. Mehta MC, Jain AC. Early repolarization on scalar electrocardiogram. Am J Med Sci 1995;309:305–311.
18. Gussak I, Antzelevitch C. Early repolarization syndrome: clinical characteristics and possible cellular and ionic mechanisms. J Electrocardiol 2000;33:299–309.
19. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation 1999;100:1660–1666.
20. Shu J, Zhu T, Yang L, Cui C, Yan GX. ST-segment elevation in the early repolarization syndrome, idiopathic ventricular fibrillation, and the Brugada syndrome: cellular and clinical linkage. J Electrocardiol 2005;38:26–32.
21. Antzelevitch C. J wave syndromes: molecular and cellular mechanisms. J Electrocardiol 2013;46:510–518.
22. Mahida S, Derval N, Sacher F et al. History and clinical significance of early repolarization syndrome. Heart Rhythm 2015;12:242–249.
23. Wellens HJ, Schwartz PJ, LindemansFWet al. Risk stratification for sudden cardiac death: current status and challenges for the futuredagger. Eur Heart J 2014;35: 1642–1651.
24. Macfarlane P, Antzelevitch C, Haissaguerre M, Huikuri HV, Potse M, Rosso R, Sacher F, Tikkanen J, Wellens H, Yan GX. The early repolarization pattern: consensus paper. J Am Coll Cardiol 2015;66:470–477.
25. Kanter RJ, Pfeiffer R, Hu D, Barajas-Martinez H, Carboni MP, Antzelevitch C. Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation 2012;125:14–22.
26. Antzelevitch C. Molecular biology and cellular mechanisms of Brugada and long QT syndromes in infants and young children. J Electrocardiol 2001;34:177–181.
27. Wedekind H, Smits JP, Schulze-Bahr E et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation 2001;104: 1158–1164.
28. Nagase S, Kusano KF, Morita H, Fujimoto Y, Kakishita M, Nakamura K, Emori T, Matsubara H, Ohe T. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol 2002;39:1992–1995.
29. Nakagawa K, Nagase S, Morita H, Ito H. Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm 2014;11:314–317.
30. Koncz I, Gurabi Z, Patocskai B, Panama BK, Szel T, Hu D, Barajas-Martinez H, Antzelevitch C. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol 2014;68C:20–28.
31. Antzelevitch C. Brugada syndrome. Pacing ClinElectrophysiol 2006;29:1130–1159.
32. Coronel R, Casini S, Koopmann TT et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005;112:2769–2777.
33. Bastiaenen R, Hedley PL, Christiansen M, Behr ER. Therapeutic hypothermia and ventricular fibrillation storm in early repolarization syndrome. Heart Rhythm 2010; 7:832–834.
34. Federman NJ, Mechulan A, Klein GJ, Krahn AD. Ventricular fibrillation induced by spontaneous hypothermia in a patient with early repolarization syndrome. J Cardiovasc Electrophysiol 2013;24:586–588.
35. Sakabe M, Fujiki A, Tani M, Nishida K, Mizumaki K, Inoue H. Proportion and prognosis of healthy people with coved or saddle-back type ST segment elevation in the right precordial leads during 10 years follow-up. EurHeart J 2003;24: 1488–1493.
36. Tsuji H, Sato T, Morisaki K, Iwasaka T. Prognosis of subjects with Brugada- type electrocardiogram in a population of middle-aged Japanese diagnosed during a health examination. Am J Cardiol 2008;102:584–587.
37. Gervacio-Domingo G, Isidro J, Tirona J, Gabriel E, David G, Amarillo ML, Morales D, Dans A. The Brugada type 1 electrocardiographic pattern is common among Filipinos. J Clin Epidemiol 2008;61:1067–1072.
38. Ito HYK, Chen R, He Q, Curb JD. The prevalence and prognosis of a Brugadatype electrocardiogram in a population of middle-aged Japanese-American men with follow-up of three decades. Am J Med Sci 2006;331:4.
39. Letsas KP,Weber R, Astheimer K, Kalusche D, Arentz T. Tpeak-Tend interval and Tpeak-Tend/QT ratio as markers of ventricular tachycardia inducibility in subjects with Brugada ECG phenotype. Europace 2010;12:271–274.
40. GallagherMM, Forleo GB, Behr ER, Magliano G, De LL, Morgia V, De LF, Romeo F. Prevalence and significance of Brugada-type ECG in 12,012 apparently healthy European subjects. Int J Cardiol 2008;130:44–48.
41. Pecini R, Cedergreen P, Theilade S, Haunso S, Theilade J, Jensen GB. The prevalence and relevance of the Brugada-type electrocardiogram in the Danish general population: data from the Copenhagen City Heart Study. Europace 2010;12: 982–986.
42. Patel SS, Anees SS, Ferrick KJ. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin Electrophysiol 2009;32: 704–708.
43. Lee C, Soni A, Tate RB, Cuddy TE. The incidence and prognosis of Brugada electrocardiographic pattern in the Manitoba Follow-Up Study. Can J Cardiol 2005;21: 1286–1290.
44. Tikkanen JT, Anttonen O, Junttila MJ, Aro AL, Kerola T, Rissanen HA, Reunanen A, Huikuri HV. Long-term outcome associated with early repolarization on electrocardiography. NEnglJ Med 2009;361:2529–2537.
45. Sinner MF, ReinhardW, Muller M et al. Association of early repolarization pattern on ECG with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA). PLoS Med 2010;7:e1000314.
46. Haruta D, Matsuo K, Tsuneto A, Ichimaru S, Hida A, Sera N, Imaizumi M, Nakashima E, Maemura K, Akahoshi M. Incidence and prognostic value of early repolarization pattern in the 12–lead electrocardiogram. Circulation 2011;123: 2931–2937.
47. Hayashi M, ShimizuW, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015;116:1887–1906.
48. Brosnan MJ, Kumar S, LaGerche A, Brown A, Stewart S, Kalman JM, Prior DL. Early repolarization patterns associated with increased arrhythmic risk are common in young non-Caucasian Australian males and not influenced by athletic status. Heart Rhythm 2015;12:1576–1583.
49. Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, Giordano U, Pappone C, Mascioli G, Rossetti G, De Nardis R, Colombo M. Risk stratification in Brugada syndrome: results of the PRELUDE (Programmed ELectrical stimUlation preDictive valuE) registry. Journal of the American College of Cardiology 2012;59:37–45.
50. Sroubek J, Probst V, Mazzanti A et al. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation 2016.
51. Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. Right ventricular electrocardiographic leads for detection of Brugada syndrome in sudden unexplained death syndrome survivors and their relatives. ClinCardiol 2001;24:776–781.
52. Miyamoto K, Yokokawa M, Tanaka K, Nagai T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, ShimizuW. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol 2007;99:53–57.
53. Nagase S, Hiramatsu S, Morita H, Nishii N, Murakami M, Nakamura K, Kusano KF, Ito H, Ohe T. Electroanatomical correlation of repolarization abnormalities in Brugada syndrome: detection of type 1 electrocardiogram in the right ventricular outflow tract. J Am Coll Cardiol 2010;56:2143–2145.
54. Veltmann C, Papavassiliu T, Konrad T, Doesch C, Kuschyk J, Streitner F, Haghi D, Michaely HJ, Schoenberg SO, Borggrefe M, Wolpert C, Schimpf R. Insights into the location of type I ECG in patients with Brugada syndrome: correlation of ECG and cardiovascular magnetic resonance imaging. Heart Rhythm 2012;9: 414–421.
55. Richter S, Sarkozy A, Paparella G, Henkens S, Boussy T, Chierchia GB, Brugada R, Brugada J, Brugada P. Number of electrocardiogram leads displaying the diagnostic coved-type pattern in Brugada syndrome: a diagnostic consensus criterion to be revised. EurHeart J 2010;31:1357–1364.
56. Veerakul G, Nademanee K. Brugada syndrome: two decades of progress. Circ J 2012;76:2713–2722.
57. Brugada P, Brugada J, Roy D. Brugada syndrome 1992–2012: 20 years of scientific excitement, and more. Eur Heart J 2013;34:3610–3615.
58. Huikuri HV, Juhani Junttila M. Clinical aspects of inherited J-wave syndromes. Trends Cardiovasc Med 2015;25:24–30.
59. Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm 2015;12:1852–1866.
60. Olde Nordkamp LR, Vink AS, Wilde AA, de Lange FJ, de Jong JS, WielingW, van Dijk N, Tan HL. Syncope in Brugada syndrome: prevalence, clinical significance, and clues from history taking to distinguish arrhythmic from nonarrhythmic causes. Heart Rhythm 2015;12:367–375.
61. Ikeda T, Abe A, Yusa S, Nakamura K, Ishiguro H, Mera H, Yotsukura M, Yoshino H. The full stomach test as a novel diagnostic technique for identifying patients at risk for Brugada Syndrome. J Cardiovasc Electrophysiol 2006;17:602–607.
62. Shimeno K, Takagi M, Maeda K, Tatsumi H, Doi A, Yoshiyama M. Usefulness of multichannel Holter ECG recording in the third intercostal space for detecting type 1 Brugada ECG: comparison with repeated 12–lead ECGs. J Cardiovasc Electrophysiol 2009;20:1026–1031.
63. Viskin S, Rosso R, Friedensohn L, Havakuk O, Wilde AA. Everybody has Brugada syndrome until proven otherwise? Heart Rhythm 2015;12:1595 1598.
64. Conte G, de Asmundis C, Ciconte G, Julia J, Sieira J, Chierchia GB, Brugada P. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA 2014;312:2039–2041.
65. Conte G, Dewals W, Sieira J et al. Drug-induced Brugada syndrome in children: clinical features, device-based management and long-termfollow up. J Am Coll Cardiol 2014;63:2272–2279.
66. Gandjbakhch E, Fressart V, Duthoit G, Marquie C, Deharo JC, Pousset F, Hebert JL, Simon F, Himbert C, Klug D, Charron P, Hidden-Lucet F. Malignant response to ajmaline challenge in SCN5A mutation carriers: experience from a large familial study. Int J Cardiol 2014;172:256–258.
67. Kligfield P, Gettes LS, Bailey JJ et al. Recommendations for the standardization and interpretation of the electrocardiogram: part I: the electrocardiogram and its technology a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society endorsed by the International Society for Computerized Electrocardiology. Journal of the American College of Cardiology 2007;49:1109–1127.
68. Papavassiliu T, Wolpert C, Fluchter S, Schimpf R, Neff W, Haase KK, Duber C, Borggrefe M. Magnetic resonance imaging findings in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2004;15:1133–1138.
69. Papavassiliu T, Veltmann C, Doesch C, Haghi D, Germans T, Schoenberg SO, van Rossum AC, Schimpf R, Brade J, Wolpert C, Borggrefe M. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm 2010;7:1790–1796.
70. Takagi M, Aihara N, Kuribayashi S, Taguchi A, Kurita T, Suyama K, Kamakura S, Takamiya M. Abnormal response to sodiumchannel blockers in patients with Brugada syndrome: augmented localised wall motion abnormalities in the right ventricular outflow tract region detected by electron beam computed tomography. Heart 2003;89:169–174.
71. Catalano O, Antonaci S, Moro G et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur- Heart J 2009;30:2241–2248.
72. van Hoorn F, Campian ME, Spijkerboer A, Blom MT, Planken RN, van Rossum AC, de Bakker JM, Wilde AA, Groenink M, Tan HL. SCN5A mutations in Brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PLoS One 2012;7:e42037.
73. Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin JA. Clinical approaches to tachyarrhythmias. The Brugada syndrome. Vol 10. Armonk, NY: Futura Publishing Company, Inc. 1999.
74. Antzelevitch C. Brugada syndrome: historical perspectives and observations. Eur- Heart J 2002;23:676–678.
75. Slezak J, Tribulova N, Okruhlicova L, Dhingra R, Bajaj A, Freed D, Singal P. Hibernating myocardium: pathophysiology, diagnosis, and treatment. Can J Physiol Pharmacol 2009;87:252–265.
76. Nademanee K, Raju H, de Noronha SV et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J Am Coll Cardiol 2015;66: 1976–1986.
77. Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/ dysplasia: clinical impact of molecular genetic studies. Circulation 2006;113:1634–1637.
78. Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011; 97:530–539.
79. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. The New England journal of medicine 1988; 318:129–133.
80. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 2010;107:700–714.
81. Delmar M, Makita N. Cardiac connexins, mutations and arrhythmias. Curr OpinCardiol 2012;27:236–241.
82. Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, BirchmeierW, Mohler P, van Veen TA, van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 2012;95:460–468.
83. Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation 2001;103:710–717.
84. Cerrone M, Lin X, Zhang M et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014;129:1092–1103.
85. Corrado D, Leoni L, Link MS et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003;108:3084–3091.
86. Matsuo K, Kurita T, Inagaki M, Kakishita M, Aihara N, Shimizu W, Taguchi A, Suyama K, Kamakura S, Shimomura K. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. EurHeart J 1999;20: 465–470.
87. Peters S, Trummel M, Denecke S, Koehler B. Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia cardiomyopathy. Int J Cardiol 2004;95:207–210.
88. Peters S, Trummel M, Koehler B. QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Heart Rhythm 2008;5:1417–1421.
89. Postema PG,Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, Priori SG, Tan HL, Hiraoka M, Brugada J, Wilde AA. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website. ,www. brugadadrugs.org.. Heart Rhythm 2009;6:1335–1341.
90. Shimizu W. Acquired forms of the Brugada syndrome. J Electrocardiol 2005;38 (Suppl):22–25.
91. Brugada P, Brugada J, Brugada R. Arrhythmia induction by antiarrhythmic drugs. Pacing Clin Electrophysiol 2000;23:291–292.
92. Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000;101:510–515.
93. Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol 1996;27:1061–1070.
94. Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol 2002;25:395–398.
95. Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med 2002;346:1591 1592.
96. Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol 2001;12: 275–275.
97. Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol 2001;34:345–349.
98. Nogami A, Nakao M, Kubota S et al. Enhancement of J-ST-segment elevation by the glucose and insulin test in Brugada syndrome. Pacing Clin Electrophysiol 2003; 26:332–337.
99. Araki T, Konno T, Itoh H, Ino H, Shimizu M. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J 2003;67:93–95.
100. Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol 2001;12: 1192–1194.
101. Chiale PA, Garro HA, Fernandez PA, Elizari MV. High-degree right bundle branch block obscuring the diagnosis of Brugada electrocardiographic pattern. Heart Rhythm 2012;9:974–976.
102. Baranchuk A, Nguyen T, Ryu MH, Femenia F, Zareba W, Wilde AA, Shimizu W, Brugada P, Perez-Riera AR. Brugada phenocopy: new classification. Ann Noninvasive Electrocardiol 2012;17:299–314.
103. Nam GB. Idiopathic ventricular fibrillation, early repolarization and other J wave-related ventricular fibrillation syndromes. Circ J 2012;76:2723 2731.
104. Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation 1996;93:372–379.
105. McIntyre WF, Perez-Riera AR, Femenia F, Baranchuk A. Coexisting early repolarization pattern and Brugada syndrome: recognition of potentially overlapping entities. J Electrocardiol 2012;45:195–198.
106. Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, Arzamendi D, Berne P, Brugada R, Brugada P, Brugada J. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol 2008;52: 1567–1573.
107. Kamakura T, Kawata H, Nakajima I et al. Significance of non-type 1 anterior early repolarization in patients with inferolateral early repolarization syndrome. J Am Coll Cardiol 2013;62:1610–1618.
108. Kawata H, Noda T, Yamada Y, Okamura H, Satomi K, Aiba T, Takaki H, Aihara N, Isobe M, Kamakura S, ShimizuW. Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and Brugada syndrome. Heart Rhythm 2012;9:77–83.
109. Matsumoto AM. Fundamental aspects of hypogonadism in the aging male. Rev Urol 2003;5(Suppl 1):S3–S10.
110. Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: a Brugada syndrome variant? J Cardiovasc Electrophysiol 2000;11:95–98.
111. Aizawa Y, Sato A, Watanabe H et al. Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J Am Coll Cardiol 2012;59:1948–1953.
112. Nademanee K. Sudden unexplained death syndrome in southeast Asia. Am J Cardiol 1997;79(6A):10–11.
113. Watanabe H, Nogami A, Ohkubo K et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol 2011;4:874–881.
114. Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST- segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
115. ShimizuW, Kamakura S. Catecholamines in children with congenital long QT syndrome and Brugada syndrome. J Electrocardiol 2001;34(Suppl):173–175.
116. Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:1277 1280.
117. Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, Ohe T, Shimizu W. Acute and chronic management in patients
with Brugada syndrome associated with electrical storm of ventricular fibrillation.
Heart Rhythm 2007;4:695–700.
118. Watanabe A, Fukushima KK, Morita H, Miura D, SumidaW, Hiramatsu S, Banba K, Nishii N, Nagase S, Nakamura K, Sakuragi S, Ohe T. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J 2006;27:1579–1583.
119. Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, Guicheney P, Di Fusco S, Rey JL, Cauchemez B, Leenhardt A. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol 2004;43:1853–1860.
120. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 2004;110:1731–1737.
121. Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2002;13:698–701.
122. Iguchi K, Noda T, Kamakura S, Shimizu W. Beneficial effects of cilostazol in a patient with recurrent ventricular fibrillation associated with early repolarization syndrome. Heart Rhythm 2013;10:604–606.
123. Agac MT, Erkan H, Korkmaz L. Conversion of Brugada type I to type III and successful control of recurrent ventricular arrhythmia with cilostazol. Arch Cardiovasc Dis 2014;107:476–478.
124. Hasegawa K, Ashihara T, Kimura H, Jo H, Itoh H, Yamamoto T, Aizawa Y, Horie M. Long-term pharmacological therapy of Brugada syndrome: is J-wave attenuation a marker of drug efficacy? Intern Med 2014;53:1523–1526.
125. Shinohara T, Ebata Y, Ayabe R, Fukui A, Okada N, Yufu K, Nakagawa M, Takahashi N. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J-wave syndromes. Heart Rhythm 2014;11: 1441–1445.
126. Haissaguerre M, Sacher F, Nogami A et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J Am Coll Cardiol 2009;53:612–619.
127. Junttila MJ, Tikkanen JT, Kentta T, Anttonen O, Aro AL, Porthan K, Kerola T, Rissanen HA, Knekt P, Huikuri HV. Early repolarization as a predictor of arrhythmic and nonarrhythmic cardiac events in middle-aged subjects. Heart Rhythm 2014;11:1701–1706.
128. Nam GB, Ko KH, Kim J, Park KM, Rhee KS, Choi KJ, Kim YH, Antzelevitch C. Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J 2010;31:330–339.
129. Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, Likittanasombat K, Bhuripanyo K, Ngarmukos T. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011;123:1270–1279.
130. Sacher F, Jesel L, Jais P, Haissaguerre M. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm 2014;11:732–734.
131. Brugada J, Pappone C, Berruezo A, Vicedomini G, Manguso F, Ciconte G, Giannelli L, Santinelli V. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ Arrhythm Electrophysiol 2015;8:1373–1381.
132. Ghosh S, Cooper DH, Vijayakumar R, Zhang J, Pollak S, Haissaguerre M, Rudy Y. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm 2010;7:534–537.
133. Zhang J, Sacher F, Hoffmayer K et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015;131:1950–1959.
134. Kowalczyk E, Kasprzak JD, Lipiec P. Giant J-wave and Brugada-like pattern in a patient with severe hypothermia. Acta Cardiol 2014;69:66–67.
135. RuDusky BM. The electrocardiogram in hypothermia-the J wave and the Brugada syndrome. Am J Cardiol 2004;93:671–672.
Rosso R, Ben-Shachar S, Antzelevitch C, Viskin S. Fever-induced Brugada pattern: how common is it and what does it mean? Heart Rhythm 2013;10:1375–1382.
137. Ansari E, Cook JR. Profound hypothermia mimicking a Brugada type ECG. J Electrocardiol 2003;36:257–260.
138. Amin AS, Meregalli PG, Bardai A, Wilde AA, Tan HL. Fever increases the risk for cardiac arrest in the Brugada syndrome. Ann Intern Med 2008;149:216–218.
139. Rattanawong P, Vutthikraivit W, Charoensri A, Jongraksak T, Prombandankul A, Kanjanahattakij N, Rungaramsin S, Wisaratapong T, Ngarmukos T. Fever- induced Brugada syndrome is more common than previously suspected: a cross- sectional study from an endemic area. Ann Noninvasive Electrocardiol 2016;21:136–141.
140. Noda T, ShimizuW, Tanaka K, Chayama K. Prominent J wave and ST segment elevation: serial electrocardiographic changes in accidental hypothermia. J Cardiovasc Electrophysiol 2003;14:223.
141. Tan HL, Meregalli PG. Lethal ECG changes hidden by therapeutic hypothermia. Lancet 2007;369:78.
142. Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nakama Y, Maruhashi T, Kagawa E, Dai K, Aokage T, Matsushita J, Ikenaga H. Therapeutic hypothermia after out-of-hospital cardiac arrest due to Brugada syndrome. Resuscitation 2008;79:332–335.
143. Patel RB, Ng J, Reddy V, Chokshi M, Parikh K, Subacius H, Sheikh-Ali AA, Nguyen T, Link MS, Goldberger JJ, Ilkhanoff L, Kadish AH. Early repolarization associated with ventricular arrhythmias in patients with chronic coronary artery disease. Circ Arrhythm Electrophysiol 2010;3:489–495.
144. Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced VT/VF in the setting of early repolarization and the protective effect of quinidine, cilostazol and milrinone. Circ Arrhythm Electrophysiol 2014;7:134–142.
145. Kawata H, Morita H, Yamada Y et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: a novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm 2013;10:1161–1168.
146. Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J 2012;76:1054–1065.
147. Kapplinger JD, Tester DJ, Alders M et al. An international compendium of mutations in the SCN5A encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46.
148. Ackerman MJ, Priori SG, Willems S et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011;8:1308–1339.
149. Park DS, Cerrone M, Morley G et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 2015;125: 403–412.
150. Szel T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol 2014;63:2037–2045.
151. Patocskai B, Szel T, Yoon N, Antzelevitch C. Cellular mechanisms underlying the fractionated and late potentials on epicardial electrograms and the ameliorative effect of epicardial radiofrequency ablation in an experimental model of Brugada syndrome. In: Program and Abstracts of the 24th Annual Upstate New York Cardiac Electrophysiology Society Meeting, November 3, 2014, Buffalo, NY, 2014Abstact 006.
152. Burashnikov E, Pfeiffer R, Barajas-Martinez H et al. Mutations in the cardiac L-type calcium channel associated J wave sydnrome and sudden cardiac death. Heart Rhythm 2010;7:1872–1882.
153. Cordeiro JM, Marieb M, Pfeiffer R, Calloe K, Burashnikov E, Antzelevitch C. Accelerated inactivation of the L-type calcium due to a mutation in CACNB2b due to a mutation in CACNB2b underlies Brugada syndrome. J Mol Cell Cardiol 2009;46: 695–703.
154. Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underline a new clinical entity characterized by ST segment elevation, short QT intervals, and sudden cardiac death. Circ Res 2006;99: 1279.
155. Gurnett CA, De WM, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron 1996;16:431–440.
156. Barajas-Martinez H, Hu D, Ferrer T et al. Molecular genetic and functional association of Bugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 2012;9:548–555.
157. Delaney JT, Muhammad R, Blair MA, Kor K, Fish FA, Roden DM, Darbar D. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 2012;14:1428–1432.
158. Medeiros-Domingo A, Tan BH, Crotti L et al. Gain-of-function mutation S422L in the KCNJ8–encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 2010;7:1466–1471.
159. Hu D, Barajas-Martinez H, Medeiros-Domingo A et al. Novel mutations in the sodium channel 2 subunit gene (SCN2B) associated with Brugada syndrome and atrial fibrillation. Circulation 2012;126:A16521.
160. Riuro H, Beltran-Alvarez P, Tarradas A et al. A missense mutation in the sodium channel ß2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013;34:961–966.
161. Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011;8:1024 1032.
162. Delpo´n E, Cordeiro JM, Noeæez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov A, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol 2008;1:209 218.
163. Olesen MS, Jensen NF, Holst AG, Nielsen JB, Tfelt-Hansen J, Jespersen T, Sajadieh A, Haunso S, Lund JT, Calloe K, Schmitt N, Svendsen JH. A novel nonsense variant in Nav1.5 cofactor MOG1 eliminates its sodium current increasing effect and may increase the risk of arrhythmias. Can J Cardiol 2011;27: 523–523.
164. Kattygnarath D, Maugenre S, Neyroud N et al. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet 2011;4:261–268.
165. Cerrone M, Lin X, Zhang M et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2013;129:1092–1103.
166. Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ, Pitt GS. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013;10:1886–1894.
167. Bezzina CR, Barc J, Mizusawa Y et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 2013;45:1044–1049.
168. Boukens BJ, Sylva M, de Gier-de VC, Remme CA, Bezzina C, Christoffels VM, Coronel R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ Res 2013;113: 137–141.
169. Hartman ME, Liu Y, Zhu WZ, Chien WM,Weldy CS, Fishman GI, Laflamme MA, Chin MT. Myocardial deletion of transcription factor CHF1/Hey2 results in altered myocyte action potential and mild conduction system expansion but does not alter conduction system function or promote spontaneous arrhythmias. FASEB J 2014;28:3007–3015.
170. Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, Park JE, Makiyama T, Horie M, Arimura T, Makita N, Kimura A. Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ J 2013;77:959–967.
171. Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2009;2:270–278.
172. Watanabe H, Koopmann TT, S. LS et al. Sodium channel ß1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 2008;118:2260–2268.
173. Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 2009;297:H1446–H1452.
174. Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta 2013;1833: 886–894.
175. Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation 2002; 105:707–713.
176. Hu D, Barajas-Martinez H, Medeiros-Domingo A et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na (v)1.5 and K(v)4.3 channel currents. Heart Rhythm 2012;9:760–769.
177. Hu D, Barajas-Martinez H, Pfeiffer R et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79.
178. Behr ER, Savio-Galimberti E, Barc J et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 2015;106:520–529.
179. Antzelevitch C. Cardiac repolarization. The long and short of it. Europace 2005; 7(Suppl 2):3–9.
180. Liu H, Chatel S, Simard C et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One 2013;8:e54131.
181. Perrin MJ, Adler A, Green S et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J wave syndrome associated with sudden cardiac death. Circ Cardiovasc Genet 2014;7:782–789.
182. Verkerk AO, Wilders R, Schulze-Bahr E et al. Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome 1. Cardiovasc Res 2005;68:441–453.
183. Wilders R, Verkerk AO. Role of the R1135H KCNH2 mutation in Brugada syndrome. Int J Cardiol 2010;144:149–151.
184. Ohno S, Zankov DP, Ding WG et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol 2011;4:352–361.
185. Boczek NJ, Ye D, Johnson EK et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ Res 2014;115:460–469.
186. Ueda K, Nakamura K, Hayashi T et al. Functional characterization of a traffickingdefective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 2004;279:27194–27198.
187. Noseworthy PA, Tikkanen JT, Porthan K et al. The early repolarization pattern in the general population clinical correlates and heritability. J Am Coll Cardiol 2011;57: 2284–2289.
188. ReinhardW, Kaess BM, Debiec R, Nelson CP, Stark K, Tobin MD, Macfarlane PW, Tomaszewski M, Samani NJ, Hengstenberg C. Heritability of early repolarization: a population-based study. Circ Cardiovasc Genet 2011;4:134–138.
189. Nunn LM, Bhar-Amato J, Lowe MD, Macfarlane PW, Rogers P, McKenna WJ, Elliott PM, Lambiase PD. Prevalence of J-point elevation in sudden arrhythmic death syndrome families. J Am Coll Cardiol 2011;58:286–290.
190. Haissaguerre M, Chatel S, Sacher F et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol 2009;20:93–98.
191. Schwartz PJ, Ackerman MJ, George AL Jr., Wilde AA. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013;62:169–180.
192. Raffan E, Semple RK. Next generation sequencing: implications for clinical practice. Br Med Bull 2011;99:53–71
193. Refsgaard L, Holst AG, Sadjadieh G, Haunso S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet 2012;20:905–908.
194. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 2009;120:1752–1760.
195. Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, Haunso S, Holst AG, Svendsen JH, Olesen MS. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol 2013;29: 1104–1109.
196. Kapplinger J, Tester D, Alders M et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46.
197. Hu D, Barajas-Martinez H, Terzic A et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol 2014;171:431–442.
198. Alfares AA, Kelly MA, McDermott G et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:8808–8888.
199. Le Scouarnec S, Karakachoff M, Gourraud JB et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet 2015;24:2757–2763.
200. Veeramah KR, Karafet TM, Wolf D, Samson RA, Hammer MF. The KCNJ8– S422L variant previously associated with J-wave syndromes is found at an increased frequency in Ashkenazi Jews. Eur J Hum Genet 2014;22:94–98.
201. Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunso S, Sadjadieh A, Winkel BG, Olesen M, Tfelt-Hansen J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 2013;84:489–495.
202. Campuzano O, Allegue C, Fernandez A, Iglesias A, Brugada R. Determining the pathogenicity of genetic variants associated with cardiac channelopathies. Sci Rep 2015;5:7953.
203. Crotti L, Marcou CA, Tester DJ, Castelletti S, Giudicessi JR, Torchio M, Medeiros-Domingo A, Simone S, Will ML, Dagradi F, Schwartz PJ, Ackerman MJ. Spectrum and prevalence of mutations Involving BrS1– through BrS12– susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J Am Coll Cardiol 2012; 60:1410–1418.
204. SongW, ShouW. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr Cardiol 2012;33:943–949.
205. Ruklisa D,Ware JS,Walsh R, Balding DJ, Cook SA. Bayesian models for syndromeand gene-specific probabilities of novel variant pathogenicity. Genome medicine 2015;7:5.
206. Juang JM, Lu TP, Lai LC et al. Utilizing multiple in silico analyses to identify putative causal SCN5A variants in Brugada syndrome. Sci Rep 2014;4:3850.
207. Walsh R, Peters NS, Cook SA, Ware JS. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet 2014;51:35–44.
208. Kapplinger JD, Giudicessi JR, Tester DJ, Callis TE, Ackerman MJ. Enhanced classification of non-synonymous single nucleotide variants in the SCN5A- encoded Nav1.5 cardiac sodium channel. Heart Rhythm 2012;9:1912.
209. Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR, Makielski JC, Wilde AA, Ackerman MJ. Enhanced classification of Brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded Nav1.5 cardiac sodium channel. Circ Cardiovasc Genet 2015;8:582–595.
210. Probst V, Wilde AA, Barc J et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet 2009;2: 552–557.
211. Matsuo K, Akahoshi M, Seto S, Yano K. Disappearance of the Brugada-type electrocardiogram after surgical castration: a role for testosterone and an explanation for the male preponderance? Pacing Clin Electrophysiol 2003;26:1151–1153.
212. Antzelevitch C. Androgens and male predominance of the Brugada syndrome phenotype. Pacing Clin Electrophysiol 2003;26:1429–1431.
213. Korte AK, Derde L, van Wijk J, Tjan DH. Sudden cardiac arrest as a presentation of Brugada syndrome unmasked by thyroid storm. BMJ Case Rep 2015;2015.
214. Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, Antzelevitch C. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol 2010;49:543–553.
215. Morita H, Zipes DP,WuJ. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm 2009;6:S34–S43.
216. HoogendijkMG, Opthof T, Postema PG, Wilde AA, de Bakker JM, Coronel R. The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol 2010;3:283–290..
217. Hoogendijk MG, Potse M, Linnenbank AC et al. Mechanism of right precordial ST-segment elevation in structural heart disease: excitation failure by current-toload mismatch. Heart Rhythm 2010;7:238–248.
218. Patocskai B, Antzelevitch C. Expert opinion on orphan drugs. Expert Opin Orphan Drugs 2015.
219. Wilde AA, Postema PG. Bringing home the bacon? The next step in cardiac sodium channelopathies. J Clin Invest 2015;125:99–101.
220. Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada syndrome: a decade of progress. Circ Res 2002;91:1114–1119.
221. Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, Aihara N, Kamakura S, Kobayashi J, Kosakai Y. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol 2002;40:330–334.
222. Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ Arrhythm Electrophysiol 2014;7:134–142.
223. Huikuri HV. Separation of benign from ,alignant J waves. Heart Rhythm 2015;12: 384–385.
224. Aizawa Y, Sato M, Kitazawa H, Aizawa Y, Takatsuki S, Oda E, Okabe M, Fukuda K. Tachycardia-dependent augmentation of “notched J waves” in a general patient population without ventricular fibrillation or cardiac arrest: Not a repolarization but a depolarization abnormality? Heart Rhythm 2015;12:376–383.
225. Badri M, Patel A, Yan G. Cellular and ionic basis of J-wave syndromes. Trends Cardiovasc Med 2015;25:12–21.
226. Morita H, Kusano KF, Miura D, Nagase S, Nakamura K, Morita ST, Ohe T, Zipes DP, Wu J. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation 2008;118:1697–1704.
227. Terho HK, Tikkanen JT, Junttila JM, Anttonen O, Kentta TV, Aro AL, Kerola T, Rissanen HA, Reunanen A, Huikuri HV. Prevalence and prognostic significance of fragmented QRS complex in middle-aged subjects with and without clinical or electrocardiographic evidence of cardiac disease. Am J Cardiol 2014;114: 141–147.
228. Osher HL, Wolff L. Electrocardiographic patern simulating acute myocardial injury. Am J Med Sci 1953;226:541–545.
229. Viskin S, Adler A, Halkin A, Rosso R. Reply: is the J wave or the ST slope malignant. . .or neither? J Am Coll Cardiol 2014;63:1812–1813.
230. Rosso R, Adler A, Halkin A, Viskin S. Risk of sudden death among young individuals with J waves and early repolarization: putting the evidence into perspective. Heart Rhythm 2011;8:923–929.
231. Aagaard P, Shulman E, Di Biase L, Fisher JD, Gross JN, Kargoli F, Kim SG, Palma EC, Ferrick KJ, Krumerman A. Prognostic value of automatically detected early repolarization. Am J Cardiol 2014;114:1431–1436.
232. Uberoi A, Jain NA, Perez M,Weinkopff A, Ashley E, Hadley D, Turakhia MP, Froelicher V. Early repolarization in an ambulatory clinical population. Circulation 2011;124:2208–2214.
233. Muramoto D, Yong CM, Singh N, Aggarwal S, Perez M, Ashley E, Hadley D, Froelicher V. Patterns and prognosis of all components of the J-wave pattern in multiethnic athletes and ambulatory patients. Am Heart J 2014;167:259–266.
234. Perez MV, Uberoi A, Jain NA, Ashley E, Turakhia MP, Froelicher V. The prognostic value of early repolarization with ST-segment elevation in African Americans. Heart Rhythm 2012;9:558–565.
235. Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med 2003;115:171–177.
236. Rollin A, Maury P, Bongard V, Sacher F, Delay M, Duparc A, Mondoly P, Carrie D, Ferrieres J, Ruidavets JB. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol 2012; 110:1302–1308.
237. Tikkanen JT, Junttila MJ, Anttonen O, Aro AL, Luttinen S, Kerola T, Sager SJ, Rissanen HA, Myerburg RJ, Reunanen A, Huikuri HV. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 2011;123:2666–2673.
238. Olson KA, Viera AJ, Soliman EZ, Crow RS, Rosamond WD. Long-term prognosis associated with J-point elevation in a large middle-aged biracial cohort: the ARIC study. Eur Heart J 2011;32:3098–3106.
239. Rosso R, Halkin A, Viskin S. J waves and early repolarization: do not confuse me with the facts! Heart Rhythm 2012;9:1603–1604.
240. Demidova MM, Martin-Yebra A, van der Pals J, Koul S, Erlinge D, Laguna P, Martinez JP, Platonov PG. Transient and rapid QRS-widening associated with a J-wave pattern predicts impending ventricular fibrillation in experimental myocardial infarction. Heart Rhythm 2014;11:1195–1201.
241. Shinde R, Shinde S, Makhale C, Grant P, Sathe S, Durairaj M, Lokhandwala Y, Di Diego JM, Antzelevitch C. Occurrence of “J waves” in 12–lead ECG as a marker of acute ischemia and their cellular basis. Pacing Clin Electrophysiol 2007;30:817–819.
242. Gourraud JB, Le Scouarnec S, Sacher F et al. Identification of large families in early repolarization syndrome. J Am Coll Cardiol 2013;61:164 172.
243. Watanabe H, Makiyama T, Koyama T et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm 2010;7:647–652.
244. Letsas KP, Charalampous C, Korantzopoulos P, Tsikrikas S, Bramos D, Kollias G, Efremidis M, Sideris A. Novel indexes of heterogeneity of ventricular repolarization in subjects with early repolarization pattern. Europace 2012;14:877–878.
245. Rosso R, Glikson E, Belhassen B, Katz A, Halkin A, Steinvil A, Viskin S. Distinguishing “benign” from “malignant early repolarization”: The value of the ST-segment morphology. Heart Rhythm 2012;9:225–229.
246. Mahida S, Derval N, Sacher F et al. Role of electrophysiological studies in predicting risk of ventricular arrhythmia in early repolarization syndrome. J Am Coll Cardiol 2015;65:151–159.
247. Probst V, Veltmann C, Eckardt L et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643.
248. Kamakura S, Ohe T, Nakazawa K et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1–V3. Circ Arrhythm Electrophysiol 2009; 2:495–503.
249. Conte G, Sieira J, Ciconte G et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: a 20-year single-center experience. J Am Coll Cardiol 2015;65: 879–888.
250. Sacher F, Probst V, Maury P et al. Outcome after implantation of a cardioverterdefibrillator in patients with Brugada syndrome: a multicenter study: part 2. Circulation 2013;128:1739–1747.
251. Priori SG, Gasparini M, Napolitano C, Della BP, Ottonelli AG, Sassone B, Giordano U, Pappone C, Mascioli G, Rossetti G, De NR, Colombo M. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed Electrical stimUlation preDictive valuE) registry. J Am Coll Cardiol 2012;59:37–45.
252. Take Y, Morita H, Toh N, Nishii N, Nagase S, Nakamura K, Kusano KF, Ohe T, Ito H. Identification of high-risk syncope related to ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm 2012;9:752–759.
253. Conte G, De Asmundis C, Sieira J, Levinstein M, Chierchia GB, DiGiovanni G, Baltogiannis G, Ciconte G, Saitoh Y, Casado-Arroyo R, Pappaert G, Brugada P. Clinical characteristics, management, and prognosis of elderly patients with Brugada syndrome. J Cardiovasc Electrophysiol 2014;25:514–519.
254. Priori SG, Napolitano C, Glordano U, Collisani G, Memmi M. Brugada syndrome and sudden cardiac death in children. Lancet 2000;355:808–809.
255. Probst V, Denjoy I, Meregalli PG et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007;115:2042–2048.
256. Meregalli PG, Tan HL, Probst V et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009;6:341–348.
257. Cordeiro JM, Barajas-Martinez H, Hong K et al. Compound heterozygous mutations P336L and I1660 V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation 2006;114:2026–2033.
258. Nunez L, Barana A, Amoros I et al. p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 2013;10:264–272.
259. Poelzing S, Forleo C, Samodell M et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 2006;114: 368–376.
260. Viswanathan PC, BensonDW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest 2003;111: 341–346.
261. Sommariva E, Pappone C, Martinelli BF, Di RC, Rosaria CM, Salvi E, Vergara P, Sala S, Cusi D, Ferrari M, Benedetti S. Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. EurJ Hum Genet 2013;21: 911–917.
262. Delise P, Allocca G, Marras E et al. Risk stratification in individuals with the Brugada type 1 ECG pattern without previous cardiac arrest: usefulness of a combined clinical and electrophysiologic approach. Eur Heart J 2011;32:169–176.
263. Yokokawa M, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, ShimizuW. Neurally mediated syncope as a cause of syncope in patients with Brugada electrocardiogram. J Cardiovasc Electrophysiol 2010;21: 186–192.
264. Richter S, Sarkozy A, Veltmann C, Chierchia GB, Boussy T,Wolpert C, Schimpf R, Brugada J, Brugada R, Borggrefe M, Brugada P. Variability of the diagnostic ECG pattern in an ICD patient population with Brugada syndrome. J Cardiovasc Electrophysiol 2009;20:69–75.
265. Veltmann C, Schimpf R, Echternach C, Eckardt L, Kuschyk J, Streitner F, Spehl S, Borggrefe M, Wolpert C. A prospective study on spontaneous fluctuations between diagnostic and non-diagnostic ECGs in Brugada syndrome: implications for correct phenotyping and risk stratification. Eur Heart J 2006;27:2544–2556.
266. Rolf S. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J 2003;24:1104–1112.
267. Veltmann C,Wolpert C, Sacher F, Mabo P, Schimpf R, Streitner F, Brade J, Kyndt F, Kuschyk J, Le MH, Borggrefe M, Probst V. Response to intravenous ajmaline: a retrospective analysis of 677 ajmaline challenges. Europace 2009;11:1345–1352.
268. Okamura H, Kamakura T, Morita H et al. Risk stratification in patients with Brugada syndrome without previous cardiac arrest: prognostic value of combined risk factors. Circ J 2015;79:310–317.
269. Govindan M, Batchvarov VN, Raju H, Shanmugam N, Bizrah M, Bastiaenen R, Kiotsekoglou A, Camm J, Behr ER. Utility of high and standard right precordial leads during ajmaline testing for the diagnosis of Brugada syndrome. Heart 2010;96:1904–1908.
270. Brugada J, Brugada R, Brugada P. Electrophysiologic testing predicts events in Brugada syndrome patients. Heart Rhythm 2011;8:1595–1597.
271. Wilde AA, Viskin S. EP testing does not predict cardiac events in Brugada syndrome. Heart Rhythm 2011;8:1598–1600.
272. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: a cardiac ticking timebomb? Europace 2007;9:707–710.
273. Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AA, Breithardt G, Eckardt L. Role of programmed ventricular stimulation in patients with Brugada syndrome: a meta-analysis of worldwide published data. Eur Heart J 2007;28: 2126–2133.
274. Brugada P, Brugada R, Brugada J. Patients with an asymptomatic Brugada electrocardiogram should undergo pharmacological and electrophysical testing. Circulation 2005;112:279–285.
275. Sieira J, Conte G, Ciconte G et al. Prognostic value of programmed electrical stimulation in Brugada syndrome: 20 years experience. Circ Arrhythm Electrophysiol 2015;8:777–784.
276. Makimoto H, Kamakura S, Aihara N et al. Clinical impact of the number of extrastimuli in programmed electrical stimulation in patients with Brugada type 1 electrocardiogram. Heart Rhythm 2012;9:242–248.
277. Tokioka K, Kusano KF, Morita H, Miura D, Nishii N, Nagase S, Nakamura K, Kohno K, Ito H, Ohe T. Electrocardiographic parameters and fatal Arrhythmic events in patients with Brugada syndrome: combination of depolarization and repolarization abnormalities. J Am Coll Cardiol 2014;63:2131–2138.
278. Take Y, Morita H. Fragmented QRS: What is the meaning? Indian Pacing Electrophysiol J 2012;12:213–225.
279. Kaneko Y, Horie M, Niwano S et al. Electrical storm in patients with Brugada syndrome is associated with early repolarization. Circ Arrhythm Electrophysiol 2014;7: 1122–1128.
280. Takagi M, Aonuma K, Sekiguchi Y, Yokoyama Y, Aihara N, Hiraoka M. Japan Idiopathic Ventricular Fibrillation Study I. The prognostic value of early repolarization (J wave) and ST-segment morphology after J wave in Brugada syndrome: multicenter study in Japan. Heart Rhythm 2013;10:533 539.
281. Rollin A, Sacher F, Gourraud JB et al. Prevalence, characteristics, and prognosis role of type 1 ST elevation in the peripheral ECG leads in patients with Brugada syndrome. Heart Rhythm 2013;10:1012–1018.
282. Marquez MF, Bisteni A, Medrano G, De Micheli A, Guevara M, Iturralde P, Colin L, Hermosillo G, Cardenas M. Dynamic electrocardiographic changes after aborted sudden death in a patient with Brugada syndrome and rate- dependent right bundle branch block. J Electrocardiol 2005;38:256 259.
283. Kasanuki H, Ohnishi S, Ohtuka M, Matsuda N, Nirei T, Isogai R, Shoda M, Toyoshima Y, Hosoda S. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 1997;95:2277–2285.
284. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997;96:2595–2600.
285. Ikeda T, Takami M, Sugi K, Mizusawa Y, Sakurada H, Yoshino H. Noninvasive risk stratification of subjects with a Brugada-type electrocardiogram and no history of cardiac arrest. Ann Noninvasive Electrocardiol 2005;10:396–403.
286. Yoshioka K, Amino M, Zareba W, Shima M, Matsuzaki A, Fujii T, Kanda S, Deguchi Y, Kobayashi Y, Ikari Y, Kodama I, Tanabe T. Identification of high- risk Brugada syndrome patients by combined analysis of late potential and T-wave amplitude variability on ambulatory electrocardiograms. Circ J 2013;77:610–618.
287. Tada T, Kusano KF, Nagase S, Banba K, Miura D, Nishii N, Watanabe A, Nakamura K, Morita H, Ohe T. Clinical significance of macroscopic T-wave alternans after sodium channel blocker administration in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2008;19:56–61.
288. Uchimura-Makita Y, Nakano Y, Tokuyama T et al. Time-domain T-wave alternans is strongly associated with a history of ventricular fibrillation in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2014;25:1021–1027.
289. Junttila MJ, Brugada P, Hong K, Lizotte E, de Zutter M, Sarkozy A, Brugada J, Benito B, Perkiomaki JS, Makikallio TH, Huikuri HV, Brugada R. Differences in 12–lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol 2008;19:380–383.
290. Maury P, Rollin A, Sacher F et al. Prevalence and prognostic role of various conduction disturbances in patients with the Brugada syndrome. Am J Cardiol 2013;11: 1384–1389.
291. Bigi MA, Aslani A, Shahrzad S. Clinical predictors of atrial fibrillation in Brugada syndrome. Europace 2007;9:947–950.
292. Makimoto H, Nakagawa E, Takaki H, Yamada Y, Okamura H, Noda T, Satomi K, Suyama K, Aihara N, Kurita T, Kamakura S, Shimizu W. Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J Am Coll Cardiol 2010;56:1576–1584.
293. Castro Hevia J, Antzelevitch C, Tornes Barzaga F, Dorantes Sanchez M, Dorticos Balea F, Zayas Molina R, Quinones Perez MA, Fayad Rodriguez Y. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol 2006;47: 1828–1834.
294. Karim Talib A, Sato N, Sakamoto N, Tanabe Y, Takeuchi T, Saijo Y, Kawamura Y, Hasebe N. Enhanced transmural dispersion of repolarization in patients with J wave syndromes. J Cardiovasc Electrophysiol 2012;23:1109–1114.
295. Lambiase PD. Tpeak-Tend interval and Tpeak-Tend/QT ratio as markers of ventricular tachycardia inducibility in subjects with Brugada ECG phenotype. Europace 2010;12:158–159.
296. Maury P, Sacher F, Gourraud JB et al. Increased Tpeak-Tend interval is highly and independently related to arrhythmic events in Brugada syndrome. Heart Rhythm 2015;12:2469–2476.
297. Sugao M, Fujiki A, Nishida K, Sakabe M, Tsuneda T, Iwamoto J, Mizumaki K, Inoue H. Repolarization dynamics in patients with idiopathic ventricular fibrillation: pharmacological therapy with bepridil and disopyramide. J Cardiovasc Pharmacol 2005;45:545–549.
298. Aizawa Y, Yamakawa H, Takatsuki S et al. Efficacy and safety of bepridil for prevention of ICD shocks in patients with Brugada syndrome and idiopathic ventricular fibrillation. Int J Cardiol 2013;168:5083–5085.
298A. Calo` L, Giustetto C, Martino A, Sciarra L, Cerrato N, Marziali M, Rauzino J, Carlino G, deRuvo E, Guerra F, Rebecchi M, Lanzillo C, Anselmino M, Castro A, Turreni F, Penco M, Volpe M, Capucci A, Gaita F. J Am Coll Cardiol 2016 Mar 29;67(12):1427–1440.
299. Masrur S, Memon S, Thompson PD. Brugada syndrome, exercise, and exercise testing. Clin Cardiol 2015;38:323–326.
300. Amin AS, de Groot EA, Ruijter JM, Wilde AA, Tan HL. Exercise-induced ECG changes in Brugada syndrome. Circ Arrhythm Electrophysiol 2009;2:531–539.
301. Brugada J, Brugada R, Brugada P. Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol 2000;33(Suppl):41–47.
302. Nademanee K, Veerakul G, Mower M, Likittanasombat K, Krittayapong R, Bhuripanyo K, Sitthisook S, Chaothawee L, Lai MY, Azen SP. Defibrillator Versus beta-Blockers for Unexplained Death in Thailand (DEBUT): a randomized clinical trial. Circulation 2003;107:2221–2226.
303. Sacher F, Probst V, Maury P et al. Outcome after implantation of cardioverter- defibrillator in patients with Brugada syndrome: a multicenter study—part 2. Circulation 2013;128:1739–1747.
304. Sacher F, Probst V, Bessouet M et al. Remote implantable cardioverter defibrillator monitoring in a Brugada syndrome population. Europace 2009;11:489–494.
305. De Maria E, Olaru A, Cappelli S. The entirely subcutaneous defibrillator (S- ICD): state of the art and selection of the ideal candidate. Curr Cardiol Rev 2015;11: 180–186.
306. Brugada P, Brugada R, Brugada J, Geelen P. Use of the prophylactic implantable cardioverter defibrillator for patients with normal hearts. Am J Cardiol 1999;83: 98D–100D.
307. Takagi M, Tatsumi H, Yoshiyama M. Approach to the asymptomatic patients with Brugada syndrome. Indian Pacing Electrophysiol J 2007;7:73 76.
308. van Den Berg MP, Wilde AA, Viersma TJW, Brouwer J, Haaksma J, van der Hout AH, Stolte-Dijkstra I, Bezzina TCR, Van Langen IM, Beaufort-Krol GC, Cornel JH, Crijns HJ. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol 2001;12:630–636.
309. Bertomeu-Gonzalez V, Ruiz-Granell R, Garcia-Civera R, Morell-Cabedo S, Ferrero A. Syncopal monomorphic ventricular tachycardia with pleomorphism, sensitive to antitachycardia pacing in a patient with Brugada syndrome. Europace 2006;8:1048–1050.
310. Lee KL, Lau C, Tse H,Wan S, Fan K. Prevention of ventricular fibrillation by pacing in a man with Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:935–937.
311. Cortez-Dias N, Placido R, Marta L, Bernardes A, Sobral S, Carpinteiro L, de Sousa J. Epicardial ablation for prevention of ventricular fibrillation in a patient with Brugada syndrome. Rev Port Cardiol 2014;33:305–305.
312. Shah AJ, Hocini M, Lamaison D, Sacher F, Derval N, Haissaguerre M. Regional substrate ablation abolishes Brugada syndrome. J Cardiovasc Electrophysiol 2011;22: 1290–1291.
313. Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3. A marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998;97:457–460.
314. Chinushi M, Aizawa Y, Ogawa Y, Shiba M, Takahashi K. Discrepant drug action of disopyramide on ECG abnormalities and induction of ventricular arrhythmias in a patient with Brugada syndrome. J Electrocardiol 1997;30:133–136.
315. Minoura Y, Di Diego JM, Barajas-Martinez H, Zygmunt AC, Hu D, Sicouri S, Antzelevitch C. Ionic and cellular mechanisms underlying the development of acquired Brugada syndrome in patients treated with antidepressants. J Cardiovasc Electrophysiol 2012;23:423–432.
316. Minoura Y, Panama BK, Nesterenko VV, Betzenhauser M, Barajas-Martinez H, Hu D, Di Diego JM, Antzelevitch C. Effect of Wenxin Keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm 2013;10:1054–1062.
317. Johnson P, Lesage A, Floyd WL, YoungWGJr, Sealy WC. Prevention of ventricular fibrillation during profound hypothermia by quinidine. Ann Surg 1960;151: 490–495.
318. Belhassen B, Shapira I, Shoshani D, Paredes A, Miller H, Laniado S. Idiopathic ventricular fibrillation: inducibility and beneficial effects of class I antiarrhythmic agents. Circulation 1987;75:809–816.
319. Belhassen B, Viskin S, Antzelevitch C. The Brugada syndrome: is an implantable cardioverter defibrillator the only therapeutic option? Pacing Clin Electrophysiol 2002;25:1634–1640.
320. Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1420–1422.
321. Belhassen B, Viskin S. Pharmacologic approach to therapy of Brugada syndrome: quinidine as an alternative to ICD therapy? In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada Syndrome: From Bench to Bedside. Oxford: Blackwell Futura; 2004:202–211.
322. Viskin S, Wilde AA, Tan HL, Antzelevitch C, Shimizu W, Belhassen B. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm 2009;6:401–404.
323. Belhassen B, Glick A, Viskin S. Excellent long-term reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fibrillation and Brugada syndrome. Pacing Clin Electrophysiol 2009;32:294–301.
324. Marquez MF, Bonny A, Hernandez-Castillo E, De SA, Gomez-Flores J, Nava S, Hidden-Lucet F, Iturralde P, Cardenas M, Tonet J. Long-term efficacy of low doses of quinidine on malignant arrhythmias in Brugada syndrome with an implantable cardioverter-defibrillator: a case series and literature review. Heart Rhythm 2013; 9:1995–2000.
325. Pellegrino PL, Di BM, Brunetti ND. Quinidine for the management of electrical storm in an old patient with Brugada syndrome and syncope. Acta Cardiol 2013; 68:201–203.
326. Probst V, Allouis M, Sacher F et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J Cardiovasc Electrophysiol 2006;17:270–275.
327. Schweizer PA, Becker R, Katus HA, Thomas D. Successful acute and long-term management of electrical storm in Brugada syndrome using orciprenaline and quinine/quinidine. Clin Res Cardiol 2010;99:467–470.
328. Marquez MF, Rivera J, Hermosillo AG, Iturralde P, Colin L, Moragrega JL, Cardenas M. Arrhythmic storm responsive to quinidine in a patient with Brugada syndrome and vasovagal syncope. Pacing Clin Electrophysiol 2005;28:870–873.
329. Viskin S, Antzelevitch C, Marquez MF, Belhassen B. Quinidine: a valuable medication joins the list of “endangered species”. Europace 2007;12:1105–1106.
330. Rosso R, Glick A, Glikson M et al. Outcome after implantation of cardioverter defribrillator in patients with Brugada syndrome: a multicenter Israeli study (ISRABRU). Isr Med Assoc J 2008;10:435–439.
331. Mok NS, Chan NY, Chi-Suen CA. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol 2004;27:821–823.
332. Belhassen B, Rahkovich M, Michowitz Y, Glick A, Viskin S. Management of Brugada syndrome: a 33-year experience using electrophysiologically-guided therapy with Class 1A antiarrhythmic drugs. Circ Arrhythm Electrophysiol 2015;6:1393–1402.
333. Bouzeman A, Traulle S, Messali A, Extramiana F, Denjoy I, Narayanan K, Marijon E, Hermida JS, Leenhardt A. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace 2014;16: 572–577.
334. Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol 2001;12:268–272.
335. Kyriazis K, Bahlmann E, van der Schalk H, Kuck KH. Electrical storm in Brugada syndrome successfully treated with orciprenaline: effect of low-dose quinidine on the electrocardiogram. Europace 2009;11:665–666.
336. ShimizuW, Matsuo K, Takagi M, Tanabe Y, Aiba T, Taguchi A, Suyama K, Kurita T, Aihara N, Kamakura S. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve- lead electrocardiograms. J Cardiovasc Electrophysiol 2000;11:396–404.
337. Tanaka H, Kinoshita O, Uchikawa S et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1293–1294.
338. Maury P, Couderc P, Delay M, Boveda S, Brugada J. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace 2004;6:130–133.
339. Maury P, Hocini M, Haissaguerre M. Electrical storms in Brugada syndrome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J 2005;5:25–34.
340. Jongman JK, Jepkes-Bruin N, Ramdat Misier AR, Beukema WP, Delnoy PP, Oude LH, Dambrink JH, Hoorntje JC, Elvan A. Electrical storms in Brugada syndrome successfully treated with isoproterenol infusion and quinidine orally. Neth Heart J 2007;15:151–155.
341. Sharif-Kazemi MB, Emkanjoo Z, Tavoosi A, Kafi M, Kheirkhah J, Alizadeh A, Sadr-Ameli MA. Electrical storm in Brugada syndrome during pregnancy. Pacing Clin Electrophysiol 2011;34:e18–e21.
342. Furniss G. Isoprenaline and quinidine to calm Brugada VF storm. BMJ Case Rep 2012;2013.
343. Roten L, Derval N, Sacher F et al. Heterogeneous response of J wave syndromes to beta-adrenergic stimulation. Heart Rhythm 2012;9:1970 1976.
344. Kanlop N, Chattipakorn S, Chattipakorn N. Effects of cilostazol in the heart. J Cardiovasc Med (Hagerstown) 2011;12:88–95.
345. Bai Y, Muqier, Murakami H, Iwasa M, Sumi S, Yamada Y, Ushikoshi H, Aoyama T, Nishigaki K, Takemura G, Uno B, Minatoguchi S. Cilostazol protects the heart against ischaemia reperfusion injury in a rabbit model of myocardial infarction: focus on adenosine, nitric oxide and mitochondrial ATP-sensitive potassium channels. Clin Exp Pharmacol Physiol 2011;38:658–665.
346. Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2006;17:210–212.
347. Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to supress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm 2013;10:1720–1727.
348. Murakami M, Nakamura K, Kusano KF et al. Efficacy of low-dose bepridil for prevention of ventricular fibrillation in patients with Brugada syndrome with and without SCN5A mutation. J Cardiovasc Pharmacol 2010;56:389–395.
349. Kang L, Zheng MQ, Morishima M,Wang Y, Kaku T, Ono K. Bepridil up- regulates cardiac Na+ channels as a long-term effect by blunting proteasome signals through inhibition of calmodulin activity. Br J Pharmacol 2009;157:404–414.
350. Fish JM,Welchons DR, Kim YS, Lee SH, Ho WK, Antzelevitch C. Dimethyl lithospermate B, an extract of danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation 2006;113:1393–1400.
351. Kanlop N, Shinlapawittayatorn K, Sungnoon R, Weerateerangkul P, Chattipakorn S, Chattipakorn N. Cilostazol attenuates ventricular arrhythmia induction and improves defibrillation efficacy in swine. Can J Physiol Pharmacol 2010; 88:422–428.
352. Endoh M, Yanagisawa T, Taira N, Blinks JR. Effects of new inotropic agents on cyclic nucleotide metabolism and calcium transients in canine ventricular muscle. Circulation 1986;73:III117–III133.
353. Rapundalo ST, Grupp I, Grupp G, Abdul MM, Solaro RJ, Schwartz A. Myocardial actions of milrinone: characterization of its mechanism of action. Circulation 1986; 73:III134–III144.
354. Atarashi H, Endoh Y, Saitoh H, Kishida H, Hayakawa H. Chronotropic effects of cilostazol, a new antithrombotic agent, in patients with bradyarrhythmias. J Cardiovasc Pharmacol 1998;31:534–539.
355. Matsui K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of pimobendan on the L-type Ca2+ current and developed tension in guinea-pig ventricular myocytes and papillary muscle: comparison with IBMX, milrinone, and cilostazol. Cardiovasc Drugs Ther 1999;13:105–113.
356. Aizawa Y, Chinushi M, Hasegawa K et al. Electrical storm in idiopathic ventricular fibrillation is associated with early repolarization. J Am Coll Cardiol 2013;62: 1015–1019.
357. Nakagawa K, Nagase S, Morita H, Ito H. Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm 2013;11:314–317.
358. Morita H, Zipes DP, Morita ST, Wu J. Temperature modulation of ventricular arrhythmogenicity in a canine tissue model of Brugada syndrome. Heart Rhythm 2007;4:188–197.
359. Probst V, Veltmann C, Eckardt L et al. Long-term prognosis of aatients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643.
360. Takagi M, Aonuma K, Sekiguchi Y, Yokoyama Y, Aihara N, Hiraoka M. The prognosticvalue of early repolarization (J wave) and ST-segment morphology after J wave in Brugada syndrome: multicenter study in Japan. Heart Rhythm 2013;10: 533–539.
361. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome. Pacing Clin Electrophysiol 2003;26:957.
362. Huang Z, Patel C, Li W, Xie Q, Wu R, Zhang L, Tang R, Wan X, Ma Y, Zhen W, Gao L, Yan GX. Role of signal-averaged electrocardiograms in arrhythmic risk stratification of patients with Brugada syndrome: a prospective study. Heart Rhythm 2009;6:1156–1162.
363. Priori SG. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 2002;105:1342–1347.
364. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003;108:3092–3096.
365. Cappato R, Furlanello F, Giovinazzo V et al. J wave, QRS slurring and ST elevation in athletes with cardiac arrest in the absence of heart disease: marker of risk, or innocent bystander? Circ Arrhythm Electrophysiol 2010;3:305–311.
366. Perez-Riera AR, Abreu LC, Yanowitz F, Barros RB, Femenia F, McIntyre WF, Baranchuk A. “Benign” early repolarization versus malignant early abnormalities: clinical-electrocardiographic distinction and genetic basis. Cardiol J 2012;19:337–346.


{kind=link}