

ThS.BS. LÊ THỊ LAN HƯƠNG
PGS.TS.BS. PHẠM NGUYỄN VINH
Bệnh viện đa khoa Tâm Anh TP. Hồ Chí Minh
(…)
6. Amyloidosis tim
Thuật ngữ amyloidosis được sử dụng để mô tả tình trạng bệnh đặc trưng bởi sự tích tụ protein amyloid ngoài tế bào. Amyloidosis có thể biểu hiện như một bệnh toàn thân hoặc khu trú. Hiện nay có 3 thể amyloidosis toàn thân thường gặp nhất bao gồm: (1) Amyloidosis chuỗi nhẹ (light chain); (2) Amyloidosis ATTR thể di truyền, gây ra bởi hơn 100 đột biến gen TTR; (3) Amyloidosis thể ATTR mắc phải (không đột biến: wild-type ATTR) thường được gọi là amyloidosis hệ thống ở người lớn tuổi (senile systemic amyloidosis) ảnh hưởng chủ yếu đến nam giới cao tuổi. Amyloid lắng đọng và tích tũy theo thời gian sẽ đưa đến amyloidosis tim, mức độ lắng đọng càng nhiều thì cơ tim càng dày, cứng và cuối cùng suy giảm chức năng bơm máu của tim ( Hình 4: A-C) [17].

Hình 4: (A) Siêu âm tim 2D ở mặt phẳng bốn buồng của bệnh nhân amyloid: phì đại thất trái đồng tâm kèm tăng hồi âm. (B) Mặt phẳng bốn buồng siêu âm: phì đại thất trái kèm dãn hai nhĩ và tràn dịch màng ngoài tim lượng ít (mũi tên trắng). (C) Sinh thiết nội mạc cơ tim: tích tụ các sợi amyloid. (D, E) Hình ảnh cine trục ngắn và hình LGE ghi nhận tổn thương dưới nội mạc lan tỏa (mũi tên đỏ). CMR: cộng hưởng từ tim; LA: nhĩ trái; LV: thất trái; RA: nhĩ phải; RV: thất phải (nguồn Srinivasan và cộng sự [14])
Trong amyloidosis thể chuỗi nhẹ (AL), bất thường tương bào có nguồn gốc từ tủy xương dẫn đến mất chức năng và tích tụ protein bất thường. Những protein chuỗi nhẹ này lắng đọng và thành amyloid tích tụ trong các mô và cơ quan của cơ thể. 90% bệnh nhân AL ghi nhận có tổn thương tim và suy tim tâm trương là biểu hiện thường gặp, nếu không điều trị thích hợp, những bệnh nhân này sẽ diễn tiến xấu đưa đến suy tim hoặc rối loạn nhịp và nhanh chóng tử vong [14].
Ngược lại, amyloidosis thể ATTR do đột biến di truyền protein Transthyretin (TTR). Chức năng của protein TTR bình thường đến khi trưởng thành, và khi những protein bất thường tích lũy dần, thường là trong độ tuổi từ 30-60, sẽ gây ra amyloidosis [17]. Một số bệnh nhân bị amyloidosis thể ATTR có biểu hiện bệnh lý thần kinh tự động, một số tổn thương tim, và đôi khi có bệnh nhân tổn thương kết hợp. So với AL, amyloidosis thể ATTR tiến triển chậm hơn và bệnh nhân không được điều trị vẫn sống nhiều năm sau biểu hiện triệu chứng ban đầu của bệnh. Trái ngược với ATTR thể đột biến, amyloidosis hệ thống ở người lớn tuổi là do sự tích tụ của một phân tử TTR ‘bình thường’ nhưng không rõ nguồn gốc. Đây là một dạng bệnh tiến triển chậm và thường ảnh hưởng đến tim của nam giới ở độ tuổi 70 hoặc 80, biểu hiện bằng các triệu chứng suy tim. Tương tự như ATTR, quá trình bệnh lành tính hơn so với bệnh nhân AL [13].
Bảng 2. Đặc điểm siêu âm tim và cộng hưởng từ tim trong chẩn đoán Amyloidosis theo ESC [27]

Ở bệnh nhân có triệu chứng suy tim, kết quả siêu âm tim phát hiện dày thất trái và ECG ghi nhận biên độ thấp (mặc dù siêu âm tim khẳng định phì đại thất trái), cần cân nhắc chẩn đoán amyloidosis tim. Tăng hồi âm trên siêu âm (với biểu hiện ‘lấp lánh’) gợi ý bệnh amyloidosis tim hơn là các nguyên nhân khác gây phì đại thất trái [14,18]. Ở bệnh nhân amyloidosis, rối loạn chức năng tâm trương và kiểu hình cơ tim hạn chế thường biểu hiện trước khi rối loạn chức năng tâm thu xảy ra. Một số trường hợp, amyloidosis có thể gây ra dày giãn thất phải, dày các van tim và phì đại cơ nhú, giãn hai nhĩ cũng như phì đại vách liên nhĩ [14]. Các nghiên cứu hiện nay ghi nhận phương pháp siêu âm đo độ biến dạng toàn thể theo trục dọc (GLS) có giá trị dự đoán vượt trội trong chẩn đoán và theo dõi amyloidosis.
Đối với cộng hưởng từ tim, hình ảnh ngấm thuốc muộn trên hình LGE có thể đánh giá được lắng đọng amyloid ở khoảng kẽ. Tổn thương LGE đặc trưng của amyloidosis là tổn thương dưới nội mạc hoặc xuyên thành lan tỏa toàn bộ cơ tim thất trái (Hình 4 D-E) [15]. Trong nghiên cứu của Vogelsberg và cs, kiểu hình phân bố LGE này được phát hiện ở 12 trong số 15 bệnh nhân amyloidosis, được xác định chẩn đoán bằng sinh thiết nội mạc cơ tim [15]. Do đó, với kiểu tổn thương LGE điển hình thì độ nhạy và độ đặc hiệu của cộng hưởng từ trong chẩn đoán amyloidosis lần lượt là 80% và 94% [15]. Gần đây, tác giản Syed và cs đã đánh giá tổn thương LGE bằng cộng hưởng từ ở 120 bệnh nhân mắc bệnh amyloidosis. Trong số 35 bệnh nhân amyloidosis tim được xác định chẩn đoán về mặt mô học, tổn thương LGE xuất hiện ở 34 bệnh nhân (chiếm 97%) [16]. Kiểu hình LGE tổn thương dưới nội mạc hoặc xuyên thành lan tỏa cơ tim thất trái là phổ biến nhất, chiếm 83% và có liên quan đến lắng đọng amyloid ở khoảng kẽ (p = 0,03). Hơn nữa, sự hiện diện và kiểu hình LGE có tương quan chặt chẽ với các biểu hiện lâm sàng, hình thái, chức năng và chỉ dấu sinh học trong tiên lượng bệnh. Do đó, cộng hưởng từ là một phương tiện chẩn đoán hình ảnh không xâm lấn giúp chẩn đoán amyloidosis tim với độ chính xác cao. Tuy nhiên, cộng hưởng từ không cho phép phân loại chính xác thể bệnh amyloidosis, mặc dù điều này rất quan trọng đối với việc điều trị tiếp theo.
Bảng 3. Một số đặc điểm phân biệt thể AL và ATTR [26]
| AL amyloidosis | ATTR amyloidosis | |
| Khối cơ thất trái | Tăng nhẹ < 100 gram/m2 | Tăng nhiều > 100 gram/m2 |
| Kiểu hình phì đại | Phì đại đồng tâm | Phì đại dạng Sigmoid |
| Bề dày vách liên thất (VLT) | Bề dày VLT trong thể AL < thể ATTR | |
| LGE | Chủ yếu là LGE dưới nội mạc
QALE < 13 điểm |
Tổn thương LGE lan tỏa và xuyên thành
QALE > 13 điểm |
| Native T1 | Native T1: AL > ATTR | |
| ECV | ECV AL < ECV ATTR (≥ 40%) | |
Trong một số nghiên cứu, các tác giả ghi nhận ở bệnh amyloidosis, mức độ tổn thương LGE ở thể ATTR nhiều hơn đáng kể so với thể AL. Các tác giả đưa ra thang điểm QALE (Query Amyloid Late Enhancement) nhằm phân biệt giữa 2 thể ATTR và AL, với ngưỡng điểm QALE ≥ 13 có thể giúp phân biệt thể ATTR và thể AL với độ nhạy là 82% và độ đăc hiệu là 76% [24,26].
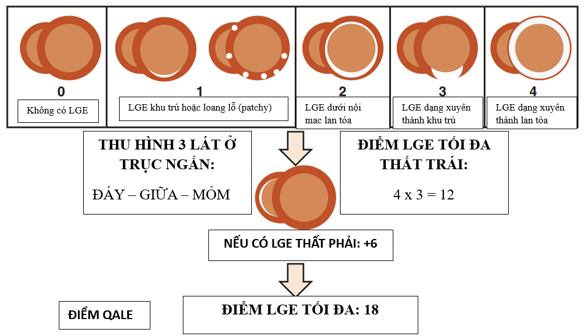
Hình 5. Thang điểm QALE trong phân biệt Amyloidosis thể AL và ATTR [26]
7. Bệnh cơ tim ty thể
Bệnh cơ tim ty thể bao gồm các dạng lâm sàng khác nhau của rối loạn thần kinh cơ với đặc điểm suy yếu chức năng chuyển hóa năng lượng của ty thể do rối loạn chức năng của chuỗi hô hấp ty thể [21]. Rối loạn ty thể có thể chỉ liên quan đến một cơ quan hoặc nhiều cơ quan khác nhau cùng một lúc và cho thấy sự khác biệt thiết yếu về mức độ nghiêm trọng của ty thể. Bệnh gây ra bởi các đột biến DNA trong nhân tế bào hoặc DNA ty thể (mtDNA). Dựa trên kiểu hình lâm sàng bệnh chia thành nhiều dạng như: liệt mắt mạn tính tiến triển (CPEO), hội chứng Kearns-Sayre (KSS), bệnh não ty thể với nhiễm toan lactic và các đợt giống đột quỵ (MELAS), hoặc động kinh được xác định bằng các sợi ragged-red (MERRF). Hiện nay, các nghiên cứu cho thấy bệnh cơ ty thể phổ biến hơn so với suy nghĩ trước đây, tần suất bệnh khoảng 1/5000 người. Các đặc điểm lâm sàng thường gặp ở rối loạn ty thể là: sụp mi, bệnh cơ gốc chi, mệt mỏi, mất khả năng gắng sức, bệnh não gan, thất điều và bệnh lý cơ tim. Một số nghiên cứu quy mô nhỏ chủ yếu dựa trên ECG và siêu âm tim đã cho thấy các dạng bệnh cơ tim khác nhau như: bệnh cơ tim phì đại hoặc bệnh cơ tim giãn nở (biểu hiện ở độ tuổi khác nhau) và tỷ lệ mắc mới dao động trong khoảng ∼15–40% [17]. Đặc biệt trong nghiên cứu của Holmgren và cs, tỷ lệ tử vong cao hơn đáng kể đã được ghi nhận ở những bệnh nhân bị bệnh cơ ty thể có tổn thương cơ tim so với những người mắc bệnh ty thể không ảnh hưởng đến tim [17]. Tử vong do tim được xác định là nguyên nhân chính gây tử vong. Do đó, việc xác định sớm những bệnh nhân mắc bệnh ty thể dễ kèm tổn thương tim rất quan trọng trên lâm sàng. Nghiên cứu của tác giả Bates và cs ở 18 bệnh nhân có cùng một đột biến mtDNA đặc hiệu có thể biểu hiện nhiều kiểu hình tim khác nhau, nhưng ngược lại tổn thương tim tương tự có thể xảy ra ở những bệnh nhân có đột biến mtDNA khác nhau. Ví dụ, kiểu hình phì đại được thường gặp trong trường hợp đột biến gen mt-tRNA (ví dụ MELAS), trong khi blốc nhĩ thất là biểu hiện thường gặp của KSS. Phì đại thất trái là kiểu hình trội của bệnh lý cơ tim gặp ở hầu hết các dạng bệnh cơ ty thể, xảy ra ở 40% số bệnh nhân này và đa phần giống bệnh cơ tim phì đại (Hình 6 A – F). Do đó, bệnh cơ ty thể là một chẩn đoán sau cùng trong trường hợp LVH không rõ nguyên nhân – đặc biệt nếu kèm theo các tổn thương ngoài tim (sụp mi, bệnh cơ gốc chi, mệt mỏi, mất khả năng gắng sức, bệnh não gan, thất điều).
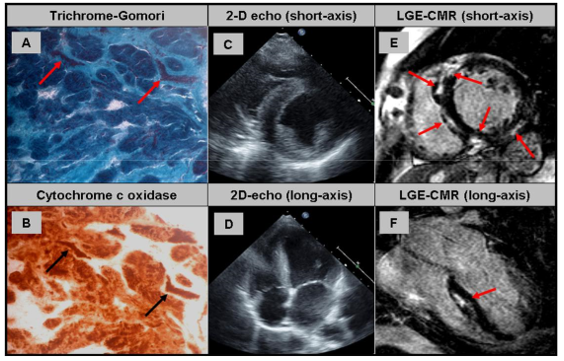
Hình 6. (A) Nhuộm trichrome-Gomori sửa đổi cho thấy sợi ‘ragged-red’ (mũi tên). (B) Phản ứng cytochrome c oxidase (tương ứng với vị trí ở hình A) xác nhận sự hiện diện của sợi “ragged-red” (mũi tên). (C + D) Siêu âm tim 2D ở mặt cắt trục ngắn và trục dài cho thấy phì đại thất trái rõ rệt. (E +F) Hình ảnh LGE trục ngắn và trục dài cho thấy: tổn thương ngấm thuốc muộn vách liên thất và vị trí ranh giới thất trái – thất phải và dạng dưới thượng mạc (sub-epi) ở thành dưới bên [23].
Các nghiên cứu đánh giá đặc điểm mô cơ tim bằng cộng hưởng từ tim sử dụng đa tham số nhằm mô tả bất thường cấu trúc ở bệnh nhân bị bệnh cơ ty thể vẫn còn hạn chế. Một nghiên cứu tiền cứu nhỏ cho thấy kiểu hình tổn thương LGE lan tỏa phân bố giữa cơ tim (intramural) ở thành dưới bên thất trái thường ghi nhận ở phân nhóm CPEO hoặc KSS [19]. Còn đối với các trường hợp bệnh nhân MELAS được báo cáo, ghi nhận kiểu hình LGE là tổn thương ngấm thuốc muộn vách liên thất và vị trí ranh giới thất trái – thất phải kéo dài thành một vệt ở vách liên thất và dạng tổn thương LGE dưới thượng mạc (sub-epi) ở thành dưới bên.
8. Các bệnh hiếm gặp khác
Bệnh dự trữ glycogen (còn gọi là mucopolysaccharidoses (MPSs)) là dạng bệnh di truyền hiếm gặp, và là bệnh lý hệ thống do rối loạn lưu trữ lysosom do sự thiếu hụt các enzyme làm suy giảm chức năng của glycosaminoglycans (GAGs). Sự lắng đọng GAGs tiến triển dẫn đến bệnh lý và suy giảm chức năng của nhiều cơ quan khác nhau. Tổn thương ở tim khá thường gặp và xuất hiện sớm trong quá trình bệnh và đặc biệt xuất hiện ở những người mắc hội chứng MPS I (Hurler), II (Hunter) và VI (Morquio). Biểu hiện ở tim, ngoài phì đại thất trái, còn đi kèm với dày và rối loạn chức năng của van tim. Tổn thương ở tim thường diễn tiến chậm, nhưng lại là một trong những nguyên nhân hàng đầu gây tử vong ở bệnh lý này [20]. Kể từ khi có các liệu pháp điều trị toàn thân như ghép tế bào gốc hoặc liệu pháp dùng enzym thay thế đã làm thay đổi tiến triển bệnh, có thể thoái triển phì đại thất trái, duy trì chức năng co bóp của tim. Vì thế, chẩn đoán sớm các trường hợp tiềm ẩn rất quan trọng trong việc điều trị, vì đây là bệnh lý có thể điều trị hồi phục. Mặt khác, vì MPS là bệnh đa cơ quan với các triệu chứng điển hình (như chậm phát triển, biến dạng xương khớp, rối loạn thần kinh mặt, liên quan đến hệ thần kinh trung ương, khiếm thị, khiếm thính, rối loạn hô hấp và rối loạn tiêu hóa) thường xảy ra sớm ở trẻ em và thanh thiếu niên, chẩn đoán chủ yếu đã được thực hiện bởi các bác sĩ nhiều chuyên ngành khác không phải là bác sĩ tim mạch. Hiện nay, các nghiên cứu cộng hưởng tim đánh giá toàn diện ở bệnh nhân MPS vẫn còn thiếu.
Tương tự như MPS, bệnh Anderson-Fabry (AFD) cũng là một rối loạn lưu trữ lysosomal, gây ra bởi khiếm khuyết men α-galactosidase dẫn đến sự tích tụ globotriaosylceramide trong khoang nội bào tiến triển của. AFD là một bệnh di truyền hiếm gặp trên nhiễn sắc thể X, do đó nam giới chủ yếu bị ảnh hưởng và biểu hiện kiểu hình lâm sàng nghiêm trọng, trong khi nữ (dạng dị hợp tử) là người mang mầm bệnh có triệu chứng hoặc không có triệu chứng của bệnh này. Biểu hiện toàn thân của AFD có thể dẫn đến tổn thương đa cơ quan gồm suy thận, lắng đọng giác mạc và các biểu hiện thần kinh, tiêu hóa và da. Kiểu hình tổn thương tim đặc trưng là phì đại thất trái giống với bệnh cơ tim phì đại hoặc bệnh tim tăng huyết áp. Nghi ngờ AFD biểu hiện ở tim trong trường hợp phì đại thất trái kèm ECG ghi nhận khoảng PR ngắn không có sóng δ và phức bộ QRS kéo dài, và rối loạn nhịp thất và rối loạn nhịp trên thất. Trong AFD, 50% bệnh nhân có tổn thương LGE trên cộng hưởng từ với vùng đáy hoặc mỏm thành dưới bên (Hình 7 A+B). Đến nay, sự phân bố xơ hóa những vùng này vẫn chưa có nguyên nhân rõ ràng [21]. Sinh thiết nội mạc cơ tim thường cho thấy các không bào (empty myocytes) trên kính hiển vi huỳnh quang và thể đặc của globotriaosylceramide trên kính hiển vi điện tử (Hình 7C). Xét nghiệm di truyền là yếu tố then chốt để chẩn đoán AFD và tương tự như các MPS khác, liệu pháp dùng enzyme thay thế có thể làm thay đổi tiến triển bệnh.
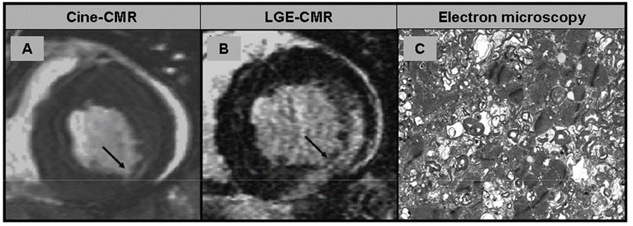
Hình 7. (A+B) Hình ảnh cine trục ngắn và hình LGE của bệnh nhân mắc bệnh Anderson-Fabry (AFD): thành dưới mỏng kèm tổn thương LGE xuyên thành. (C) Kính hiển vi điện tử thể hiện các vật thể lamellar đồng tâm đặc trưng cho bệnh lý Anderson-Fabry [2,20].
9. Kết luận
Kiểu hình phì đại thất trái có thể là kết quả của nhiều tình trạng bệnh lý khác nhau, có thể bắt nguồn từ chính cơ tim hoặc là biểu hiện một phần của bệnh hệ thống. Chẩn đoán nguyên nhân phì đại thất trái cần có một cách tiếp cận toàn diện, có hệ thống và lựa chọn từng bước các công cụ chẩn đoán thích hợp. Nền tảng của chẩn đoán chính xác bao gồm hỏi bệnh sử, tiền sử bệnh nhân, tập trung vào các bệnh lý ngoài tim và tiền sử gia đình, ghi ECG 12 chuyển đạo, theo dõi Holter và siêu âm tim. Ngoài ra, cộng hưởng từ tim với đặc điểm đánh giá mô cơ tim không xâm lấn, có thể giúp phân loại các dạng bệnh cơ tim khác nhau. Cuối cùng, sinh thiết mô cơ tim với các phân tích mô bệnh học và/hoặc phân tích di truyền sẽ cần thiết trong nhiều trường hợp, ngoài giá trị chẩn đoán còn giá trị trong điều trị và theo dõi bệnh.
TÀI LIỆU THAM KHẢO
- Maron MS. Clinical utility of cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson 2012;14:13.
- Yilmaz A, Kindermann I, Kindermann M, et al. Comparative evaluation of left and right ventricular endomyocardial biopsy:differences in complication rate and diagnostic performance. Circulation 2010;122:900–9.
- Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol 2008;51:1369–74.
- Waterhouse DF, Ismail TF, Prasad SK, et al. Imaging focal and interstitial fibrosis with cardiovascular magnetic resonance in athletes with left ventricular hypertrophy: implications for sporting participation. Br J Sports Med 2012;46(Suppl 1):i69–77.
- Maron BJ, Pelliccia A, Spirito P. Cardiac disease in young trained athletes. Insights into methods for distinguishing athlete’s heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy. Circulation 1995;91:1596–601.
- Petersen SE, Selvanayagam JB, Francis JM, et al. Differentiation of athlete’s heart from pathological forms of cardiac hypertrophy by means of geometric indices derived from cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2005;7:551–8.
- Raman SV. The hypertensive heart. An integrated understanding informed by imaging. J Am Coll Cardiol 2010;55:91–6.
- Yilmaz A, Sechtem U. Angina pectoris in patients with normal coronary angiograms: current pathophysiological concepts and therapeutic options. Heart 2012;98:1020–9.
- Rudolph AA, Abdel-Aty H, Bohl S, et al. Noninvasive detection of fibrosis applying contrast-enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy relation to remodeling. J Am Coll Cardiol 2009;53:284–91.
- Puntmann VO, Jahnke C, Gebker R, et al. Usefulness of magnetic resonance imaging to distinguish hypertensive and hypertrophic cardiomyopathy. Am J Cardiol 2010;106:1016–22.
- Noureldin RA, Liu S, Nacif MS, et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2012;14:17.
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol 2011;58:e212–60.
- Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009;120:1203–12.
- Srinivasan G, Joseph M, Selvanayagam JB. Recent advances in the imaging assessment of infiltrative cardiomyopathies. Heart 2013;99:204–13.
- Vogelsberg H, Mahrholdt H, Deluigi CC, et al. Cardiovascular magnetic resonance in clinically suspected cardiac amyloidosis: noninvasive imaging compared to endomyocardial biopsy. J Am Coll Cardiol 2008;51:1022–30
- Syed IS, Glockner JF, Feng D, et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 2010;3:155–64.
- Holmgren D, Wahlander H, Eriksson BO, et al. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J 2003;24:280–8.
- Bates MG, Bourke JP, Giordano C, et al. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J 2012;33:3023–33.
- Yilmaz A, Gdynia HJ, Ponfick M, et al. Cardiovascular magnetic resonance imaging (CMR) reveals characteristic pattern of myocardial damage in patients with mitochondrial myopathy. Clin Res Cardiol 2012;101:255–61.
- Braunlin EA, Harmatz PR, Scarpa M, et al. Cardiac disease in patients with mucopolysaccharidosis: presentation, diagnosis and management. J Inherit Metab Dis 2011;34:1183–97.
- 21 Moon JC, Sheppard M, Reed E, et al. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson 2006;8:479–82.
- Mentias, A, Raeisi-Giglou, P, Smedira, N. et al. Late Gadolinium Enhancement in Patients With Hypertrophic Cardiomyopathy and Preserved Systolic Function. J Am Coll Cardiol. 2018 Aug, 72 (8) 857–870.
- Yilmaz A, Sechtem U. Diagnostic approach and differential diagnosis in patients with hypertrophied left ventriclesHeart 2014;100:662-671.
- Jason N. Dungu et al; CMR-Based Differentiation of AL and ATTR Cardiac Amyloidosis, JACC: Cardiovascular Imaging, Volume 7, Issue 2-2014, Pages 133-142,ISSN 1936-878X
- 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol 2020;Nov 20
- Dungu JN, Valencia O, Pinney JH, Gibbs SD, Rowczenio D, Gilbertson JA, et al. CMRbased differentiation of AL and ATTR cardiac amyloidosis. JACC Cardiovasc Imaging. 2014;7(2):133–42
- Pablo Garcia-Pavia, Claudio Rapezzi, et al, Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases, European Heart Journal, Volume 42, Issue 16, 21 April 2021, Pages 1554–1568.


{kind=link}