

7.8. Các rối loạn thần kinh cơ
TS PHẠM HỮU VĂN
|
Khuyến cáo cho rối loạn thần kinh cơ Tư liệu ủng hộ khuyến cáo được tóm tắt trong tư liệu hỗ trợ online 38 |
||
|
COR |
LOE |
Các khuyến cao |
|
I |
B-NR |
1. Ở các bệnh nhân rối loạn thần kinh cơ, ICDs phòng ngừa tiên phát và thứ phát được khuyến cáo cho các chỉ định tương tự như đối với bệnh nhân có NICM nếu sống sót có ý nghĩa > 1 năm được dự kiến (1, 2). |
|
IIa |
B-NR |
2. Ở các bệnh nhân có chứng loạn dưỡng cơ Emery-Dreifuss và Limb -girdle type IB với sự liên quan đến tim tiến triển, ICD là phù hợp nếu sống sót có ý nghĩa > 1 năm được dự kiến (3-8). |
|
IIa |
B-NR |
3. Ở những bệnh nhân bị chứng loạn dưỡng cơ, theo dõi sự phát triển liên quan đến tim là hợp lý, ngay cả khi bệnh nhân không có biểu hiện triệu chứng (9-12). |
|
IIb |
B-NR |
4. Ở các bệnh nhân bị chứng loạn dưỡng cơ type 1 với chỉ định máy tạo nhịp vĩnh viễn, ICD có thể được xem xét để giảm thiểu nguy cơ SCA từ VT nếu sống sót > 1 năm được dự kiến (9, 13, 14). |
Tóm tắt
Loạn dưỡng cơ là một nhóm các bệnh di truyền ảnh hưởng đếncơxương và cơ tim. Một số biểu hiện trước tiên như NICM (ví dụ: Duchenne, Becker, và limb – girdle type 2C, 2F, và 2I), trong khi những bệnh khác biểu hiện trước tiên như thoái hóa hệ thống dẫn truyền với sự kết hợp thay đổi với bệnh cơ tim (ví dụ, chứng loạn dưỡng cơ type 1 và 2, Emery-Dreifuss, limb-girdle 1B, được tóm tắt trong Bảng 9) (15). Do SCD có thể xảy ra hoặc do VA hoặc do nhịp chậm do sự tiến triển nhanh chóng và không thể dự đoán được của bệnh hệ thống dẫn truyền, bác sĩ lâm sàng đang phải đối mặt với thách thức xác định những bệnh nhân sẽ được hưởng lợi từ máy tạo nhịp dự phòng hoặc cấy ICD. Nên có một mức độ quan tâm cao đối với những bệnh nhân bị chứng loạn dưỡng cơ có biểu hiện triệu chứng loạn nhịp (15). Các hướng dẫn hiện nay tập trung vào VA và chỉ định cho cấy ICD. Các chỉ định cho máy tạo nhịp vĩnh viễn được thảo luận trong hướng dẫn khác của ACC / AHA / HRS (16).
Văn bản Hỗ trợ Riêng biệt Khuyến cáo
1. Nói chung, chỉ định ICD ở bệnh nhân bị chứng loạn dưỡng cơ nên tuân thủ các khuyến cáo ICD chuẩn cho bệnh nhân có NICM (xem Phần 7.2.1 về Phòng ngừa Thứ phát và Mục 7.2.2 về Phòng ngừa SCD tiên phát cho NICM). Một chỉ số cao nghi ngờ về nhịp nhanh vào lại nhánh bó được bảo đảm ở các bệnh nhân có loạn dưỡng cơ có biểu hiện nhịp nhanh phức bộ QRS rộng và các triệu chứng liên quan đến nhịp nhanh (2).
2. Ở những bệnh nhân có chứng loạn dưỡng cơ Emery-Dreifuss và limb-girdle type 1B liên quan đến sự biến đổi Lamin A / C, SCD chiếm khoảng 1/3 số ca tử vong (4). Các nghiên cứu quan sát cho thấy một tỷ lệ điều trị ICD phù hợp một cách có ý nghĩa ở các bệnh nhân có rối loạn dẫn truyền tim có gene dương tính với đột biến Lamin A/C thậm chí nều chức năng LV được bảo tồn (3, 5, 17). Trong một nghiên cứu quan sát, trong đó 38% bệnh nhân bị tổn thương cơ xương đơn thuần, nhưng được bao gồm bệnh nhân có khiếm khuyết dẫn truyền và các yếu tố nguy cơ khác (bao gồm khoảng PR > 240 ms, block nhánh bó trái, NSVT, hoặc nhịp tim chậm đòi hỏi máy tạo nhịp vĩnh viễn) là tương đối phổ biến; với 52% bệnh nhân được điều trị ICD phù hợp, trong đó có khoảng 40% bệnh nhân có LVEF ≥ 45% (3). Một nghiên cứu trên các bệnh nhân có đột biến Lamin A / C, trong đó khoảng 21% có kiểu hình loạn dưỡng cơ xương, SCD và điều trị ICD phù hợp được kết hợp với NSVT, LVEF < 45%, nam giới, và đột biến Lamin A / C không phải missense 4). Các nghiên cứu quan sát này hỗ trợ việc sử dụng một ICD khi có chỉ định tạo nhịp và cũng có thể khi có bằng chứng liên quan đến tim tiến triển, NSVT hoặc LVEF giảm hiện diện.
Có rất ít dữ liệu liên quan đến hình thái hiếm của loạn dưỡng cơ Emery-Dreifuss có liên quan đến liên kết x (liên quan đến đột biến gen Emerin), nhưng loạn nhịp có thể ít gặp hơn so với đột biến Lamin A / C (15).
3. Sự liên quan đến tim có thể xảy ra ở một số loạn dưỡng thần kinh cơ (Bảng 9). Để xác định sự liên quan đến tim, ECG 12 chuyển đạo và siêu âm tim rất quan trọng cho việc đánh giá lâm sàng khởi đầu, độc lập với tình trạng triệu chứng. Nói chung, mức liên quan đến tim càng rộng, gồm các bằng chứng bệnh dẫn truyền đầu xa, rối loạn chức năng thất, và rối loạn nhịp nhĩ, thì càng nhiều khả năng VA sẽ xẩy ra. Việc đánh giá khởi đầu cho bệnh nhân loạn dưỡng cơ gồm theo dõi lưu động. Ở những bệnh nhân không triệu chứng, một số chuyên gia ủng hộ việc theo dõi hàng năm trong giai đoạn ẩn giấu của bệnh với điện tâm đồ 12 chuyển đạo hàng năm để phát hiện các bất thường dẫn truyền. Tuy nhiên, tần số tối ưu của sàng lọc điện tâm đồ còn chưa được biết rõ (18). Một khi sự liên quan đến tim xảy ra, hoặc trên cơ sở chậm dẫn truyền, loạn nhịp nhĩ, hoặc rối loạn chức năng tâm thất, một ngưỡng thấp để phát hiện các triệu chứng hoặc các phát hiện điện tim của bác sĩ lâm sàng để xác định nhu cầu phải cấy máy tạo nhịp, nghiên cứu điện sinh lý xâm lấn, hoặc cấy ICD là tối ưu.
4. Khoảng 1/3 số ca tử vong ở các bệnh nhân loạn dưỡng cơ là đột ngột (9). Mặc dù thường được cho do block dẫn truyền và vô tâm thu, SCD do VT / VF đã được ghi nhận ra ở các bệnh nhân có máy tạo nhịp vĩnh viên chức năng, và VA tự phát đã được ghi nhận ở một số trường hợp (13, 19). Nguy cơ SCD ở bệnh nhân với máy tạo nhịp cho thấy một ICD có thể được ưa chuộng hơn máy tạo nhịp tim. Tuy nhiên, các bệnh nhân này cũng có nguy cơ cao cho suy hô hấp như một nguyên nhân tranh chấp gây tử vong. Do đó, ở những bệnh nhân bị rối loạn cơ xương nặng, máy tạo nhịp hoặc ICD có thể không cải thiện hậu quả (15). Một cách tiếp cận đưa ra quyết định chia sẻ để lựa chọn ICD hoặc điều trị tạo nhịp được đảm bảo. So với bệnh nhân loạn dưỡng cơ type 1, bệnh nhân loạn dưỡng cơ type 2 không được nghiên cứu kỹ lưỡng nhưng cũng có thể có lợi từ cách tiếp cận tương tự.
Bảng 9. Rối loạn Thần kinh cơ kết hợp với Bệnh tim
|
Loạn dưỡng cơ |
Di truyền |
Gene/ Protein bị ảnh hưởng |
Bệnh lý tim tiên phát |
Tần số liên quan đến tim |
Các nguyên nhân gây tử vong |
Được kết hợp với Đột tử? |
|
Duchenne |
X-linked tạo ra kiểu hình |
Dystrophin |
NICM |
>90% |
Hô hấp, HF |
Đúng, căn nguyên chưa chắc chắn |
|
Becker |
X-linked tạo ra kiểu hình |
Dystrophin |
NICM |
60%–75% |
HF, hô hấp |
Đúng, căn nguyên chưa chắc chắn |
|
Limb-girdle type 1B |
Autosomal dominant |
Lamin A/C |
Bệnh hệ thống dẫn truyền và NICM |
>90% |
Đột ngột, HF |
Đúng |
|
Limb-girdle type 2C-2F |
Autosomal tạo ra kiểu hình |
Sarcoglycan |
NICM |
<25% |
Hô hấp, HF |
Không chắc chắn |
|
Limb-girdle type 2I |
Autosomal recessive |
Fukutin-được kiên hệ protein |
NICM |
20%–80% |
Respiratory, HF |
Không chắc chắn |
|
Myotonic type 1 |
Autosomal chiếm ưu thế |
CTG lắp lại mở rộng |
Bệnh hệ thống dẫn truyền và NICM |
60%–80% |
Hô hấp, đột ngột, HF |
30% tử vong, không chắc chắn, nhịp chậm đối lại với nhịp nhanh |
|
Myotonic type 2 |
Autosomal chiếm ưu thế |
CCTG lắp lại mở rộng |
Bệnh hệ thống dẫn truyền |
10%–25% |
Các nguyên nhân bình thường |
Đã được thống báo |
|
Emery-Dreifuss |
X-linked và autosomal chiếm ưu thế hoặc tạo ra kiểu hình |
Emerin, Lamin A/C |
Bệnh hệ thống dẫn truyền và NICM |
>90% |
Đột ngột, HF |
Đúng |
|
Facioscapulohumeral |
Autosomal chiếm ưu thế |
D4Z4 nhắc lại co bóp |
Có khả năng bệnh dẫn truyền |
5%–15% |
Các nguyên nhân bình thường, hô hấp hiếm gặp |
Chưa được thông báo |
HF: suy tim; NICM: bệnh cơ tim không do thiếu máu.
Phù hợp với sự cho phép của Groh, và cộng sự. (15).
Tài liệu tham khảo
1. Bhakta D, Groh MR, Shen C, et al. Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1. Am Heart J. 2010;160:1137-41, 41.
2. Merino JL, Carmona JR, Fernandez-Lozano I, et al. Mechanisms of sustained ventricular tachycardia in myotonic dystrophy: implications for catheter ablation. Circulation. 1998;98:541-6.
3. Anselme F, Moubarak G, Savoure A, et al. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm. 2013;10:1492-8.
4. van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol. 2012;59:493-500.
5. Meune C, Van Berlo JH, Anselme F, et al. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med. 2006;354:209-10.
6. Pasotti M, Klersy C, Pilotto A, et al. Long-term outcome and risk stratification in dilated cardiolaminopathies. J Am Coll Cardiol. 2008;52:1250-60.
7. Van Berlo JH, de Voogt WG, van der Kooi AJ, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J Mol Med. 2005;83:79-83.
8. Russo V, Nigro G. ICD role in preventing sudden cardiac death in Emery-Dreifuss muscular dystrophy with preserved myocardial function: 2013 ESC guidelines on cardiac pacing and cardiac resynchronization therapy. Europace. 2015;17:337.
9. Groh WJ, Groh MR, Saha C, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688-97.
10.Lallemand B, Clementy N, Bernard-Brunet A, et al. The evolution of infrahissian conduction time in myotonic dystrophy patients: clinical implications. Heart. 2012;98:291-6.
11.Nazarian S, Wagner KR, Caffo BS, et al. Clinical predictors of conduction disease progression in type I myotonic muscular dystrophy. Pacing Clin Electrophysiol. 2011;34:171-6.
12.Tanawuttiwat T, Wagner KR, Tomaselli G, et al. Left ventricular dysfunction and conduction disturbances in patients with myotonic muscular dystrophy type I and II. JAMA Cardiology. 2017;2:225-8.
13.Bhakta D, Shen C, Kron J, et al. Pacemaker and implantable cardioverter-defibrillator use in a US myotonic dystrophy type 1 population. J Cardiovasc Electrophysiol. 2011;22:1369-75.
14.Laurent V, Pellieux S, Corcia P, et al. Mortality in myotonic dystrophy patients in the area of prophylactic pacing devices. Int J Cardiol. 2011;150:54-8.
15.Groh WJ. Arrhythmias in the muscular dystrophies. Heart Rhythm. 2012;9:1890-95.
16.Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation. 2008;117:e350-408.
17.Kumar S, Baldinger SH, Gandjbakhch E, et al. Long-term arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. J Am Coll Cardiol. 2016;68:2299-307.
18.Brembilla-Perrot B, Luporsi JD, Louis S, et al. Long-term follow-up of patients with myotonic dystrophy: an electrocardiogram every year is not necessary. Europace. 2011;13:251-7.
19.Ha AH, Tarnopolsky MA, Bergstra TG, et al. Predictors of atrio-ventricular conduction disease, long-term outcomes in patients with myotonic dystrophy types I and II. Pacing Clin Electrophysiol. 2012;35:1262-9.
7.9. Các bệnh kênh tim
|
Các khuyến cáo cho các bệnh kênh tim Tài liệu tham khảo ủng hộ khuyến cáo được tóm tắt trong Tư liệu Hỗ trợ online 39. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở những người thân thế hệ đầu tiên của bệnh nhân có đột biến gây ra đối với hội chứng QT dài, nhịp nhanh đa hình do tăng tiết catecholamine, hội chứng QT ngắn, hoặc hội chứng Brugada, nên tư vấn di truyền và xét nghiệm di truyền đặc hiệu về đột biến được khuyến cáo (1-6). |
|
I |
B-NR |
2. Ở những bệnh nhân bệnh kênh ion tim và SCA, ICD được khuyến cáo nếu sống sót có ý nghĩa > 1 năm được dự kiến (7-13). |
Tóm tắt
Cấy ICD ở các bệnh nhân không triệu chứng nguy cơ thấp với bệnh kênh tim đối với bệnh sử gia đình SCD dương tính như chỉ định duy nhất không được ủng hộ bằng các tư liệu đã xuất bản (13-18).
Văn bản Hỗ trợ riêng biết – khuyến cáo
1. Khám lâm sàng các người thân thế hệ thứ nhất của bệnh nhân có hội chứng loạn nhịp di truyền rất quan trọng để xác định các thành viên trong gia đình bị ảnh hưởng. Do tăng nguy cơ các biến cố tim bất lợi ở bệnh nhân có kiểu gene dương tính với hội chứng QT dài, nhịp nhanh thất đa hình do tăng tiết catecholamine, và hội chứng Brugada, sàng lọc nhắm mục tiêu đột biến đặc hiệu cho gia đình được xác định có thể nhận biết các cá nhân có nguy cơ mắc các kết cục bất lợi này (5). Các xét nghiệm ECG có thể không đủ để chẩn đoán, do ECG lúc nghỉ bỉnh thường ở những bệnh nhân nhịp nhanh thất đa hình do tăng tiết catecholamine, và khoảng 25% bệnh nhân có kiểu gene dương tính với hội chứng QT dài có khoảng QTc ≤ 440 ms (2). Do tăng nguy cơ các biến cố tim bất lợi ở những bệnh nhân trẻ có hội chứng QT dài và nhịp nhanh thất đa hình do tăng tiết catecholamine (2, 19-22), sàng lọc trẻ sơ sinh và trẻ nhỏ đặc biệt quan trọng để hướng dẫn trị liệu và thiết lập các biện pháp phòng ngừa, bao gồm việc tránh các thuốc kích thích (www.crediblemeds.org) (23). Tuy nhiên, do có tới 15% đột biến liên quan đến nhịp nhanh thất đa hình do tăng tiết catecholamine dường như không gây ra bệnh (24) nên thận trọng cần tránh những điều trị không cần thiết hoặc hạn chế về thể thao đối với những cá thể đột biến nhịp nhanh đa hình dạng đột biến kiểu hình. Đáng chú ý, một số bệnh nhân có thể không muốn thực hiện thử nghiệm di truyền, do đó, tư vấn di truyền nên tập trung vào vấn đề này.
2. Bệnh nhân bệnh kênh tim (chẳng hạn, hội chứng QT dài, nhịp nhanh thất đa hình do tăng tiết catecholaminergic, hội chứng Brugada, hội chứng tái cực sớm và hội chứng QT ngắn) và SCA trước đó có nguy cơ SCA hoặc SCD tiếp theo tăng lên đáng kể (7-13, 28). Việc cấy ghép ICD làm giảm nguy cơ tử vong ở những bệnh nhân có nguy cơ cao (9, 29-31). Điều trị ICD phù hợp cho VF / VT nhanh được thông báo ở 8% đến 33% bệnh nhân kênh, trong khi các sốc không thích hợp và các biến chứng của thiết bị được báo cáo ở 8% đến 35% (10, 29, 30, 32-36). Để giảm thiểu các sốc không thích hợp, đồng thời các thuốc chẹn beta ở các bệnh nhân hội chứng QT dài và nhịp nhanh thất đa hình do tăng tiết cathecholamine, lập trình thiết bị tối ưu, và lựa chọn điện cực phù hợp là cần thiết. Tạo nhịp thất không cấy ICD được kết hợp với nguy cơ đáng kể SCA hoặc SCD tái phát ở các bệnh nhân hội chứng QT dài (37-39). Ở những bệnh nhân LQT1 được lựa chọn có SCA xảy ra không được điều trị thuốc ức chế beta khi đó liệu pháp chẹn beta có thể thay thế cho ICD ở những bệnh nhân từ chối cấy ICD (40).
Tài liệu tham khảo
1. Bai R, Napolitano C, Bloise R, et al. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol. 2009;2:6-15.
2. Goldenberg I, Horr S, Moss AJ, et al. Risk for life-threatening cardiac events in patients with kiểu gene-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51-9.
3. Nannenberg EA, Sijbrands EJ, Dijksman LM, et al. Mortality of inherited arrhythmia syndromes: insight into their natural history. Circ Cardiovasc Genet. 2012;5:183-9.
4. Priori SG, Gasparini M, Napolitano C, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59:37-45.
5. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
6. Wilde AA, Moss AJ, Kaufman ES, et al. Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation. 2016;134:872-82.
7. Wedekind H, Burde D, Zumhagen S, et al. QT interval prolongation and risk for cardiac events in kiểu gened LQTS-index children. Eur J Pediatr. 2009;168:1107-15.
8. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616-23.
9. Zareba W, Moss AJ, Daubert JP, et al. Implantable cardioverter defibrillator in high-risk long QT syndrome patients. J Cardiovasc Electrophysiol. 2003;14:337-41.
10.Monnig G, Kobe J, Loher A, et al. Implantable cardioverter-defibrillator therapy in patients with congenital long-QT syndrome: a long-term follow-up. Heart Rhythm. 2005;2:497-504.
11.Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426-34.
12.Gehi AK, Duong TD, Metz LD, et al. Risk stratification of individuals with the Brugada electrocardiogram: a meta-analysis. J Cardiovasc Electrophysiol. 2006;17:577-83.
13.Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada syndrome registry. Circulation. 2010;121:635-43.
14.Delise P, Allocca G, Marras E, et al. Risk stratification in individuals with the Brugada type 1 ECG pattern without previous cardiac arrest: usefulness of a combined clinical and electrophysiologic approach. Eur Heart J. 2011;32:169-76.
15.Hiraoka M, Takagi M, Yokoyama Y, et al. Prognosis and risk stratification of young adults with Brugada syndrome. J Electrocardiol. 2013;46:279-83.
16.Kaufman ES, McNitt S, Moss AJ, et al. Risk of death in the long QT syndrome when a sibling has died. Heart Rhythm. 2008;5:831-6.
17.Kimbrough J, Moss AJ, Zareba W, et al. Clinical implications for affected parents and siblings of probands with long-QT syndrome. Circulation. 2001;104:557-62.
18.Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342-7.
19.Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249-54.
20.Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-4.
21.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-74.
22.Spazzolini C, Mullally J, Moss AJ, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54:832-37.
23.Credible meds. Available at: http://www.crediblemeds.org. Accessed December 26, 2016.
24.Paludan-Muller C, Ahlberg G, Ghouse J, et al. Integration of 60,000 exomes and ACMG guidelines question the role of catecholaminergic polymorphic ventricular tachycardia-associated variants. Clin Genet. 2017;91:63-72.
25.Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol. 2011;58:587-95.
26.Mahida S, Derval N, Sacher F, et al. Role of electrophysiological studies in predicting risk of ventricular arrhythmia in early repolarization syndrome. J Am Coll Cardiol. 2015;65:151-9.
27.Mazzanti A, Kanthan A, Monteforte N, et al. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014;63:1300-8.
28.Siebermair J, Sinner MF, Beckmann BM, et al. Early repolarization pattern is the strongest predictor of arrhythmia recurrence in patients with idiopathic ventricular fibrillation: results from a single centre long-term follow-up over 20 years. Europace. 2016;18:718-25.
29.Etheridge SP, Sanatani S, Cohen MI, et al. Long QT syndrome in children in the era of implantable defibrillators. J Am Coll Cardiol. 2007;50:1335-40.
30.Horner JM, Kinoshita M, Webster TL, et al. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: a single-center experience. Heart Rhythm. 2010;7:1616-22.
31.Proclemer A, Ghidina M, Facchin D, et al. Use of implantable cardioverter-defibrillator in inherited arrhythmogenic diseases: data from Italian ICD Registry for the years 2001-6. Pacing Clin Electrophysiol. 2009;32:434-45.
32.Adler A, Sadek MM, Chan AY, et al. Patient outcomes from a specialized inherited arrhythmia clinic. Circ Arrhythm Electrophysiol. 2016;9:e003440.
33.Conte G, Sieira J, Ciconte G, et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: a 20-year single-center experience. J Am Coll Cardiol. 2015;65:879-88.
34.Olde Nordkamp LR, Postema PG, Knops RE, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13:443-54.
35.Rodriguez-Manero M, Sacher F, de Asmundis C, et al. Monomorphic ventricular tachycardia in patients with Brugada syndrome: a multicenter retrospective study. Heart Rhythm. 2016;13:669-82.
36.Rosso R, Glick A, Glikson M, et al. Outcome after implantation of cardioverter defibrillator [corrected] in patients with Brugada syndrome: a multicenter Israeli study (ISRABRU). Isr Med Assoc J. 2008;10:435-9.
37.Dorostkar PC, Eldar M, Belhassen B, et al. Long-term follow-up of patients with long-QT syndrome treated with beta-blockers and continuous pacing. Circulation. 1999;100:2431-6.
38.Eldar M, Griffin JC, Van Hare GF, et al. Combined use of beta-adrenergic blocking agents and long-term cardiac pacing for patients with the long QT syndrome. J Am Coll Cardiol. 1992;20:830-7.
39.Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84:1524-9.
40.Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215-21.
7.9.1. Các hội chứng bệnh kênh tim chuyên biệt
7.9.1.1. Hội chứng QT dài bẩm sinh
|
Các khuyến cáo cho hội chứng QT dài Tài liệu tham khảo ủng hộ các khuyến cáo được tóm tắt trong Tài liệu Hỗ trợ Online 40. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân hội chứng QT dài với QTc lúc nghỉ lớn hơn 470 ms, nên dùng thuốc chẹn beta (1-5). |
|
I |
B-NR |
2. Ở các bệnh nhân hội chứng QT dài có triệu chứng có nguy cơ cao thuốc ức chế beta không có hiệu quả hoặc không dung nạp, tăng cường điều trị với các thuốc bổ xung (được hướng dẫn bằng cách xem xét type hội chứng QT dài riêng biệt), bóc bỏ giao cảm tim bên trái và / hoặc ICD được khuyến cáo (2, 6-12). |
|
I |
B-NR |
3. Ở các bệnh nhân có hội chứng QT dài và các sốc ICD thích hợp mặc dù liều dung nạp tối đa của một thuốc chẹn beta, tăng cường điều trị bằng thuốc bổ sung (được hướng dẫn bằng cách xem xét type hội chứng QT dài riêng biệt) hoặc bóc bỏ giao cảm tim trái, được khuyến cáo (6, 7, 10, 13-16). |
|
I |
B-NR |
4. Ở những bệnh nhân có hội chứng QT dài được chẩn đoán lâm sàng, nên tư vấn di truyền và xét nghiệm di truyền được khuyến cáo (17-21). |
|
IIa |
B-NR |
5. Ở các bệnh nhân nghi ngờ hội chứng QT dài, theo dõi điện tâm đồ lưu động, ghi lại ECG khi nằm và ngay lập tức khi đứng, và / hoặc thực hiện test gắng sức thảm lăn có thể hữu ích cho việc tính toán chẩn đoán và theo dõi đáp ứng đối với điều trị (22-29). |
|
IIa |
B-NR |
6. Ở các bệnh nhân hội chứng QT dài không triệu chứng và QTc khi nghỉ < 470 ms, điều trị kéo dài với thuốc chẹn beta là hợp lý (3, 30, 31). |
|
IIb |
B-NR |
7. Ở các bệnh nhân hội chứng QT dài không có triệu chứng và QTc khi nghỉ > 500 ms trong khi tiếp nhận thuốc chẹn beta, tăng cường điều trị bằng thuốc (được hướng dẫn bằng cách xem xét type hội chứng QT dài riêng biệt), bóc bỏ giao cảm tim trái hoặc ICD có thể được xem xét (2, 8, 11, 30). |
|
III: Harm |
B-NR |
8. Ở các bệnh nhân có hội chứng QT kéo dài, thuốc làm kéo dài QT có thể gây nguy hại (5, 12, 32-34). |
Bảng 10 và Hình 9, 10, 11, và 12
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
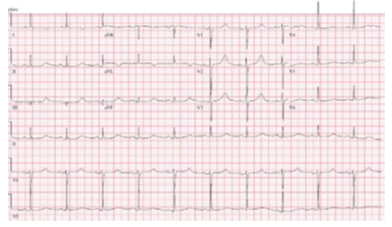
1. Beta blockers làm giảm các biến cố tim bất lợi đối với hội chứng QT dài type 1 (Hình 10) (> 95%), hội chứng QT dài type 2 (Hình 11) (> 75%), và nữ giới có hội chứng QT dài type 3 (Hình 12 ) bằng > 60% (1-5). Có tư liệu còn hạn chế về hiệu quả của beta blockers ở nam giới có hội chứng QT dài type 3 (3, 35, 36), nhưng ở những bệnh nhân được lựa chọn, beta blockers có thể bảo vệ chống lại SCA (36, 37). Một số nghiên cứu quan sát đã báo cáo hiệu quả giảm nguy cơ trong hội chứng QT kéo dài với propranolol, atenolol và nadolol với liều dùng thích hợp (26, 28, 38-40), trong khi metoprolol có hiệu quả thấp hơn (41). RCTs để đánh giá hiệu quả so sánh của các thuốc chẹn beta cụ thể còn chưa có, mặc dù nhiều trung tâm thích sử dụng nadolol. Đối với hội chứng QT dài type 1, 1 nghiên cứu báo cáo atenolol làm giảm nguy cơ VA, trong khi nadolol không liên quan đến giảm nguy cơ (2). Đối với hội chứng QT dài type 2, nadolol được báo cáo là có hiệu quả cao hơn (1, 2). Bệnh nhân dùng thuốc chẹn beta cần được theo dõi liên tục để đánh giá sự thay đổi về QTc theo thời gian và mức độ ức chế beta với gắng sức (26, 28)
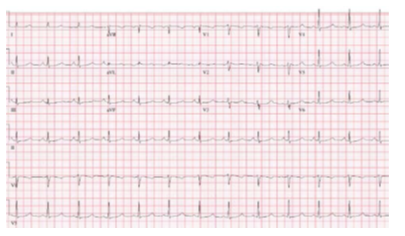
2. Các bệnh nhân có QTc dài có nguy cơ cao gồm những người có QTc > 500 ms, kiểu gene hội chứng QT dài type 2 và hội chứng QT dài type 3, nữ giới có kiểu gene hội chứng QT dài type 2, < 40 tuổi, khởi phát triệu chứng khi < 10 tuổi, và các bệnh nhân ngưng tim từ trước hoặc ngất tái phát (3, 8, 11, 30, 38). Nữ giới có hội chứng QT dài type 2 có nguy cơ ngừng tim / SCD sau sinh cao hơn (42, 43) và nên được tư vấn trước khi có thai. Bệnh nhân có hội chứng QT dài và ngất tái phát trong khi nhận được beta blockers có nguy cơ SCA gia tăng hoặc các sốc ICD phù hợp (9) và việc leo thang điều trị để ngăn ngừa SCD. Các nghiên cứu trước đây đã báo cáo lợi ích của việc đo nồng độ kháng thể, với ngất tái phát hoặc ngừng tim đã báo cáo ở 7% đến 24% bệnh nhân (44-47). Ở những bệnh nhân có nguy cơ cao, các nghiên cứu quan sát hỗ trợ hiệu quả của ICD trong việc ngăn ngừa SCD, với việc xem xét bóc bỏ giao cảm tim trái để giảm tần số sốc ICD (16, 48, 49). Bóc bỏ giao cảm tim trái có thể giảm gánh nặng VA, nhưng lên tới 27% bệnh nhân có nguy cơ cao trải qua ít nhất 1 lần tái phát (16,48,50). Bóc bỏ giao cảm tim trái có thể hiệu quả hơn ở các bệnh nhân có hội chứng QT dài type 1 và hội chứng QT dài type 3 (16). Các biến chứng liên quan đến bóc bỏ giao cảm tim trái xảy ra ở 8% đến 20% bệnh nhân (48, 51). Ngất ở những bệnh nhân có hội chứng QT dài có thể xảy ra do ngất phế vị, không tuân thủ với thuốc, hoặc chứng nhịp cùng với thuốc cùng lúc (5). Đánh giá lâm sàng kết hợp việc xem xét kiểu gen, khoảng QTc, tuân thủ điều trị thuốc và chia sẻ quyết định liên quan đến sự cần thiết phải thay đổi hoặc leo thang liệu pháp là rất quan trọng. Sử dụng thuốc bổ sung được hướng dẫn bằng type hội chứng QT dài. Trong hội chứng QT dài type 3 ranolazine 3, mexiletine, và flecainide làm rút ngắn QTc và đã được sử dụng để giảm loạn nhịp tái phát (6, 7, 10).
3. Mexiletine là một loại thuốc bổ sung có thể dùng cho bệnh nhân có hội chứng QT dài và các cơn sốc ICD tái phát. Bóc bỏ giao cảm tim bên trái được kết hợp với giảm số các sốc ICD phù hợp và gánh nặng VA (13-16). Giảm QTc < 500 ms sau khi bóc bỏ giao cảm tim trái có tương quan với giảm nguy cơ sốc ICD tái phát và tần số các triệu chứng (16, 52); tuy nhiên, SCD hoặc SCA được báo cáo ở 3% đến 10% bệnh nhân (15, 16, 48, 50). Mặc dù gánh nặng rối loạn nhịp thường giảm, có tới 27% bệnh nhân có nguy cơ cao gặp ít nhất 1 lần tái phát (13, 14, 48). Kết quả bệnh nhân được cải thiện nếu bóc bỏ giao cảm tim trái được thực hiện ở các trung tâm chuyên sâu ngoại khoa về thủ thuật này. Sử dụng thuốc bổ sung được hướng dẫn bằng type hội chứng QT dài. Trong nhóm hội chứng QT dài type 3, ranolazyn, mexiletin, và flecainide rút ngắn QTc và đã được sử dụng để giảm loạn nhịp tái phát (6, 7, 10).
4. Kiểm tra di truyền các đột biến gây bệnh trong hội chứng QT kéo dài cung cấp thông tin chẩn đoán, tiên lượng và điều trị quan trọng thêm vào đánh giá lâm sàng, và test dương tính có thể dễ dàng tính toán nguy cơ cho các thành viên trong gia đình. Năng suất xét nghiệm di truyền ở các bệnh nhân kiểu hình dương tính từ 50% đến 86%, có biểu hiện phạm vi cao hơn với sự kéo dài QT hoặc bệnh sử gia đình SCD dương tính (17, 21, 53). Xét nghiệm di truyền âm tính không loại trừ chẩn đoán hội chứng QT kéo dài, điều này dựa vào đánh giá lâm sàng. Ở các bệnh nhân không triệu chứng với QTc ≥ 480 ms kéo dài không giải thích được là trên một loạt các ECG, xét nghiệm di truyền có thể giúp khẳng định chẩn đoán và bổ sung thông tin tiên lượng thêm vào các triệu chứng lâm sàng và khoảng thời gian QTc (5, 18-20, 30, 35, 54-56).
5. Trong một nghiên cứu quan sát tiền cứu bệnh nhân nghi ngờ hội chứng QT dài, bệnh nhân có tiền sử ngất hoặc ngưng tim và hoặc người thân thế hệ thứ nhất bị ảnh hưởng hoặc giới hạn hoặc khoảng QTc kéo dài đã được thực hiện test gắng sức treadmill và test gắng sức xe đạp, với ECG được ghi trước đó, trong và sau khi gắng sức, cũng như ở các vị trí khác nhau (27). Hội chứng QT kéo dài đã được khẳng định bằng xét nghiệm di truyền ở tất cả các cá nhân bị ảnh hưởng. Trong số những bệnh nhân có khoảng QTc lúc nghỉ từ đường giới hạn đến bình thường, kéo dài thời gian phục hồi 4 phút QTc ≥ 445 ms có độ nhạy cao để xác định đúng bệnh nhân có hội chứng QT dài (27). Một nghiên cứu ở những bệnh nhân trẻ tuổi đã chứng minh QTc kéo dài > 460 ms ở 7 phút hồi phục dự đoán hội chứng QT dài type 1 hoặc bệnh nhân QT type 2 so với nhóm chứng (23). Trong một nghiên cứu sử dụng bài tập đạp xe đạp, bệnh nhân có hội chứng QT dài tiềm tàng (latent) có tăng lên trong QTc với gắng sức lớn hơn một cách có ý nghĩa so với hoặc nhóm chứng hoặc những người có kéo dài QTc lúc ban đầu (24). Những phát hiện này có thể hữu ích trong việc xác định có hội chứng QT dài. Theo dõi sự phù hợp của liệu pháp chẹn beta bằng cách sử dụng test gắng sức có thể có lợi, đặc biệt ở bệnh nhân ở tuổi học đường (26,28). Điều trị chẹn beta có thể kết hợp với giảm khi không hoạt động và QTc đỉnh cao khi gắng sức, với ngoại trừ ở bệnh nhân hội chứng QT dài type 1 với các đột biến vòng lặp C (25).
6. Khoảng 10% đến 36% bệnh nhân gene dương tính với hội chứng QT dài có khoảng QTc ≤ 440 ms, phần lớn bệnh nhân có hội chứng QT dài type 1 (31, 35). Các bệnh nhân có hội chứng QT dài và QTc bình thường có nguy cơ VA và SCD thấp hơn so với những người có QTc kéo dài (35), nhưng vẫn có nguy cơ cao về SCA hoặc SCD so với gene âm tính theo độ tuổi, và giới tính bệnh nhân (31). Thuốc chẹn beta làm giảm nguy cơ các biến cố tim bất lợi đáng kể (1-5, 30, 36, 38, 41, 57). Trong những giai đoạn có nguy cơ cao nhất trong 3 thập niên đầu của cuộc đời (11, 18), điều trị với thuốc chẹn beta có thể làm giảm nguy cơ SCA (26, 28, 36, 38). Sự thay đổi QTc xảy ra theo thời gian, đặc biệt là trong giai đoạn dậy thì và trong và sau khi mang thai, cho thấy nhu cầu đánh giá QTc trên ECG hàng năm hoặc với sự thay đổi thuốc, và đánh giá hiệu quả của thuốc với test gắng sức là khả thi. Các bệnh nhân người lớn không triệu chứng với các khoảng QTc bình thường có thể chọn Các bệnh nhân QT mãn tính không triệu chứng (QTc) có QTc bình thường có thể chọn điều trị theo hướng beta-blocker (11, 34).
7. Nguy cơ các biến cố tim bất lợi từ VA đã bị ảnh hưởng do các khoảng QTc khi nghỉ của bệnh nhân, tuổi tác, giới tính và kiểu gene / đột biến của hội chứng QT dài. Đối với nam giới không triệu chứng có hội chứng QT dài, nguy cơ xảy ra các biến cố tim mạch cao nhất trong thời thơ ấu (2, 8, 11, 30) trong thời gian tuân thủ thuốc là một thách thức. Phụ nữ trẻ có LQT2 và QTc> 500 ms có nguy cơ gia tăng chứng SCA (2, 11, 18-20, 30, 35) đặc biệt trong 9 tháng sau đẻ và có thể là ứng cử viên cho việc đặt ICD ngăn ngừa tiên phát hoặc sử dụng áo khử rung tim (30).
8. Nguy cơ các phản ứng phụ tăng lên ở những bệnh nhân có hội chứng QT kéo dài với việc kéo dài QTc> 500 ms (2, 12, 26, 35, 41, 58). Không nên dùng thuốc kéo dài QT (www.crediblemeds.org) (59) ở bệnh nhân có hội chứng QT dài, trừ không có sự thay thế phù hợp; cần theo dõi cẩn thận QTc trong khi điều trị được khuyến cáo, với việc xem xét ngưng dùng thuốc với sự kéo dài QTc đáng kể. Việc sử dụng đồng thời các thuốc kích thích hoặc suy giảm sự chú ý các thuốc không kích thích / các thuốc hoạt động quá mức được kết hợp với nguy cơ ngất / ngừng tim tăng lên trong hội chứng QT dài, đặc biệt ở nam giới, trong 1 nghiên cứu (34), nhưng dường như không liên quan đến nguy cơ gia tăng trong nghiên cứu hồi cứu khác (60). Các đợt torsades de pointes có thể bị thúc đẩy bằng bộc lộ đối với các thuốc kéo dài QT, hoặc giảm kali máu được tạo ra bằng thuốc lợi tiểu hoặc do tiếp xúc với thuốc kéo dài của QT, hoặc giảm kali máu do thuốc lợi tiểu hoặc các thuốc giảm đau dạ dầy ruột. Chú ý đến việc duy trì sự cân bằng kali và magiê bình thường khi dùng thuốc hoặc tình huống làm thúc đẩy cạn kiệt đã được gặp là một thành phần quan trọng trong điều chỉnh. Các báo cáo trường hợp hiếm gặp tồn tại khoảng QT kéo dài do sốt ở các bệnh nhân có hội chứng QT dài type 2; sốt nên được giảm bằng các thuốc hạ sốt (61) (Bảng 10).
Bảng 10. Các thuốc kéo dài QT được sử dụng thông thường (59, 62)
|
Các mẫu thuốc kéo dài QT * |
|||
|
Các thuốc chống loạn nhịp |
Các thuốc hướng tâm thần |
Các kháng sinh |
Các thuốc khác |
|
Disopyramide Procainamide (N-acetylprocainamide) Quinidine Dofetilide Dronedarone Ibutilide Sotalol Amiodarone† |
Haloperidol Phenothiazines Citalopram Tricyclic antidepressants |
Erythromycin Pentamidine Azithromycin Chloroquine Ciprofloxacin Fluconazole Levofloxacin Moxifloxacin Clarithromycin Itraconazole Ketoconazole |
Methadone Probucol Droperidol Ondansetron |
*Danh sách đầy đủ hơn được thấy trong: www.crediblemeds.org (59).
†Amiodarone hiếm khi gây torsades de pointes.
Hình 9. Ngăn ngừa SCD ở các bệnh nhân có hội chứng QT dài
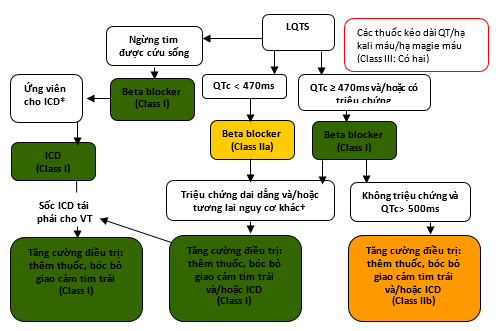
Màu sắc tương ứng với Class khuyến cáo ở bảng 1.
Xem phần 7.9.1.1 để thảo luận.
*Ứng viên ICD khi được xác định bằng trạng thái chức năng, tuổi thọ, hoặc ưa chuộng của bệnh nhân.
†Các bệnh nhân nguy cơ cao với LQTS gồm QTc > 500 ms, kiểu genes LQT2 và LQT3, nữ giới với kiểu gene LQT2, < 40 tuổi, khởi đầu các triệu chứng ở <10 tuổi, và các bệnh nhân có ngất tái phát.
ICD: máy khử rung tim có thể cấy; LQTS: hội chứng QT dài, VT: nhịp nhanh thất.
Hình 10. Hội chứng QT dài Long Type 1
Hình 11. Hội chứng QT dài Type 2
Tài liệu tham khảo
1. Abu-Zeitone A, Peterson DR, Polonsky B, et al. Efficacy of different beta-blockers in the treatment of long QT syndrome. J Am Coll Cardiol. 2014;64:1352-8.
2. Goldenberg I, Bradley J, Moss A, et al. Beta-blocker efficacy in high-risk patients with the congenital long-QT syndrome types 1 and 2: implications for patient management. J Cardiovasc Electrophysiol. 2010;21:893-901.
3. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616-23.
4. Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329-37.
5. Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long-QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215-21.
6. Chorin E, Hu D, Antzelevitch C, et al. Ranolazine for congenital long-qt syndrome type III: experimental and long-term clinical data. Circ Arrhythm Electrophysiol. 2016;9:e004370.
7. Chorin E, Taub R, Medina A, et al. Long-term flecainide therapy in type 3 long QT syndrome. Europace. 2017;euw439 [Epub ahead of print].
8. Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249-54.
9. Jons C, Moss AJ, Goldenberg I, et al. Risk of fatal arrhythmic events in long QT syndrome patients after syncope. J Am Coll Cardiol. 2010;55:783-8.
10.Mazzanti A, Maragna R, Faragli A, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol. 2016;67:1053-8.
11.Nannenberg EA, Sijbrands EJ, Dijksman LM, et al. Mortality of inherited arrhythmia syndromes: insight into their natural history. Circ Cardiovasc Genet. 2012;5:183-9.
12.Wedekind H, Burde D, Zumhagen S, et al. QT interval prolongation and risk for cardiac events in kiểu gened LQTS-index children. Eur J Pediatr. 2009;168:1107-15.
13.Collura CA, Johnson JN, Moir C, et al. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752-9.
14.Hofferberth SC, Cecchin F, Loberman D, et al. Left thoracoscopic sympathectomy for cardiac denervation in patients with life-threatening ventricular arrhythmias. J Thorac Cardiovasc. Surg. 2014;147:404-9.
15.Schneider HE, Steinmetz M, Krause U, et al. Left cardiac sympathetic denervation for the management of life-threatening ventricular tachyarrhythmias in young patients with catecholaminergic polymorphic ventricular tachycardia and long QT syndrome. Clin Res Cardiol. 2013;102:33-42.
16.Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long-QT syndrome. Circulation. 2004;109:1826-33.
17.Bai R, Napolitano C, Bloise R, et al. Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ Arrhythm Electrophysiol. 2009;2:6-15.
18.Costa J, Lopes CM, Barsheshet A, et al. Combined assessment of sex- and mutation-specific information for risk stratification in type 1 long QT syndrome. Heart Rhythm. 2012;9:892-8.
19.Kim JA, Lopes CM, Moss AJ, et al. Trigger-specific risk factors and response to therapy in long QT syndrome type 2. Heart Rhythm. 2010;7:1797-805.
20.Migdalovich D, Moss AJ, Lopes CM, et al. Mutation and gender-specific risk in type 2 long QT syndrome: implications for risk stratification for life-threatening cardiac events in patients with long QT syndrome. Heart Rhythm. 2011;8:1537-43.
21.Tester DJ, Will ML, Haglund CM, et al. Effect of clinical kiểu hình on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006;47:764-8.
22.Adler A, van der Werf C, Postema PG, et al. The phenomenon of “QT stunning”: The abnormal QT prolongation provoked by standing persists even as the heart rate returns to normal in patients with long QT syndrome. Heart Rhythm. 2012;9:901-8.
23.Aziz PF, Wieand TS, Ganley J, et al. Kiểu gene- and mutation site-specific QT adaptation during exercise, recovery, and postural changes in children with long-QT syndrome. Circ Arrhythm Electrophysiol. 2011;4:867-73.
24.Chattha IS, Sy RW, Yee R, et al. Utility of the recovery electrocardiogram after exercise: a novel indicator for the diagnosis and genotyping of long QT syndrome? Heart Rhythm. 2010;7:906-11.
25.Laksman ZW, Hamilton RM, Chockalingam P, et al. Mutation location effect on severity of kiểu hình during exercise testing in type 1 long-QT syndrome: impact of transmembrane and C-loop location. J Cardiovasc Electrophysiol. 2013;24:1015-20.
26.Moltedo JM, Kim JJ, Friedman RA, et al. Use of a cardioselective beta-blocker for pediatric patients with prolonged QT syndrome. Pediatr Cardiol. 2011;32:63-6.
27.Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124:2187-94.
28.Villain E, Denjoy I, Lupoglazoff JM, et al. Low incidence of cardiac events with beta-blocking therapy in children with long QT syndrome. Eur Heart J. 2004;25:1405-11.
29.Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955-61.
30.Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-4.
31.Goldenberg I, Horr S, Moss AJ, et al. Risk for life-threatening cardiac events in patients with kiểu gene-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51-9.
32.Choy AM, Lang CC, Chomsky DM, et al. Normalization of acquired QT prolongation in humans by intravenous potassium. Circulation. 1997;96:2149-54.
33.Kannankeril P, Roden DM, Darbar D. Drug-induced long QT syndrome. Pharmacol Rev. 2010;62:760-81.
34.Zhang C, Kutyifa V, Moss AJ, et al. Long-QT syndrome and therapy for attention deficit/hyperactivity disorder. J Cardiovasc Electrophysiol. 2015;26:1039-44.
35.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866-74.
36.Wilde AA, Moss AJ, Kaufman ES, et al. Clinical aspects of type 3 long-QT syndrome: an international multicenter study. Circulation. 2016;134:872-82.
37.Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm. 2009;6:113-20.
38.Garson A Jr, Dick M, Fournier A, et al. The long QT syndrome in children. An international study of 287 patients. Circulation. 1993;87:1866-72.
39.Priori SG, Bossaert LL, Chamberlain DA, et al. ESC-ERC recommendations for the use of automated external defibrillators (AEDs) in Europe. Eur Heart J. 2004;25:437-45. 40.Steinberg C, Padfield GJ, Al-Sabeq B, et al. Experience with bisoprolol in long-QT1 and long-QT2 syndrome. J Interv Card Electrophysiol. 2016;47:163-70.
41.Chockalingam P, Crotti L, Girardengo G, et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: higher recurrence of events under metoprolol. J Am Coll Cardiol. 2012;60:2092-9.
42.Khositseth A, Tester DJ, Will ML, et al. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60-4.
43.Rashba EJ, Zareba W, Moss AJ, et al. Influence of pregnancy on the risk for cardiac events in patients with hereditary long QT syndrome. LQTS Investigators. Circulation. 1998;97:451-6.
44.Dorostkar PC, Eldar M, Belhassen B, et al. Long-term follow-up of patients with long-QT syndrome treated with beta-blockers and continuous pacing. Circulation. 1999;100:2431-6.
45.Eldar M, Griffin JC, Van Hare GF, et al. Combined use of beta-adrenergic blocking agents and long-term cardiac pacing for patients with the long QT syndrome. J Am Coll Cardiol. 1992;20:830-7.
46.Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84:1524-9.
47.Viskin S, Glikson M, Fish R, et al. Rate smoothing with cardiac pacing for preventing torsade de pointes. Am J Cardiol. 2000;86:111k-5k.
48.Bos JM, Bos KM, Johnson JN, et al. Left cardiac sympathetic denervation in long QT syndrome: analysis of therapeutic nonresponders. Circ Arrhythm Electrophysiol. 2013;6:705-11.
49.Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-9.
50.Li J, Liu Y, Yang F, et al. Video-assisted thoracoscopic left cardiac sympathetic denervation: a reliable minimally invasive approach for congenital long-QT syndrome. Ann Thorac Surg. 2008;86:1955-8.
51.De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation. 2015;131:2185-93.
52.Li C, Hu D, Shang L, et al. Surgical left cardiac sympathetic denervation for long QT syndrome: effects on QT interval and heart rate. Heart Vessels. 2005;20:137-41.
53.Burns C, Ingles J, Davis AM, et al. Clinical and genetic features of Australian families with long QT syndrome: a registry-based study. J Arrhythm. 2016;32:456-61.
54.Barsheshet A, Goldenberg I, Uchi J, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation. 2012;125:1988-96.
55.Crotti L, Spazzolini C, Schwartz PJ, et al. The common long-QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366-75.
56.Zhang C, Kutyifa V, McNitt S, et al. Identification of low-risk adult congenital LQTS patients. J Cardiovasc Electrophysiol. 2015;26:853-8.
57.Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494-9.
58.Spazzolini C, Mullally J, Moss AJ, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54:832-7.
59.Credible meds. Available at: http://www.crediblemeds.org. Accessed December 26, 2016.
60.Rohatgi RK, Bos JM, Ackerman MJ. Stimulant therapy in children with attention-deficit/hyperactivity disorder and concomitant long QT syndrome: a safe combination? Heart Rhythm. 2015;12:1807-12.
61.Amin AS, Klemens CA, Verkerk AO, et al. Fever-triggered ventricular arrhythmias in Brugada syndrome and type 2 long-QT syndrome. Neth Heart J. 2010;18:165-9.
62.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013-22.
7.9.1.2. Nhịp nhanh thất đa hình do tiết Catecholamine
|
Các khuyến cáo cho nhịp nhanh thất đa hình do tăng tiết cathecholamine Tài liệu tham khảo ủng hộ các khuyến cáo được tóm tắt trong tư liệu hỗ trợ online 41. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân nhịp nhanh thất đa hình do tiết catecholamine, beta blocker được khuyến cáo (1, 2). |
|
I |
B-NR |
2. Ở các bệnh nhân nhịp nhanh thất đa hình do tiết catecholamine và VT dai dẳng tái phát hoặc ngất, trong khi dùng beta blocker phù hợp hoặc chịu đựng tối đa, điều trị tăng cường với hoặc điều trị phối hợp (như beta blocker, flecainide), bóc bỏ giao cảm tim trái, và / hoặc ICD được khuyến cáo (2-6). |
|
IIa |
B-NR |
3. Ở các bệnh nhân nhịp nhanh thất đa hình do tiết cathecholamine và VT trên lâm sàng hoặc ngất khi gắng sức, tư vấn di truyền và test di truyền là phù hợp (7). |
Văn bản hỗ trợ riêng biệt khuyến cáo
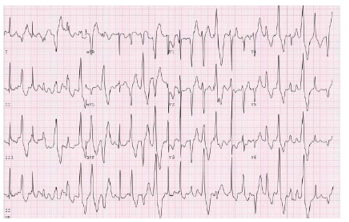
1. Nhịp nhanh thất đa hình do tiết catecholamine được đặc trưng bằng VT đa hình liên quan đến gắng sức hoặc VT hai chiều (bidirectional) (hình 13), được kết hợp với ngất và SCA. SCA / SCD được báo cáo từ 3% đến 13% bệnh nhân (1, 2, 8). Điều trị bằng thuốc beta blockers với việc giảm đi trong các biến cố tim bất lợi (1, 2). Một số chuyên gia thích sử dụng nadolol so với các thuốc chẹn beta khác; dữ liệu so sánh trực tiếp giữa các thuốc chẹn bêta không có sẵn. Việc sử dụng liều tối đa được dung nạp của thuốc beta blockers rất quan trọng. Các nghiên cứu quan sát nhỏ cho thấy có thể có lợi ích của các thuốc chẹn kênh calcium nondihydropyridine trong điều trị nhịp nhanh thất đa hình do tiết catecholamine (9, 10).
2. Flecainide kết hợp với thuốc chẹn beta có thể ngăn chặn ngoại vị thất đạt đến 76% ở những bệnh nhân nhịp nhanh thất đa hình do tiết catecholamine trong quá trình test gắng sức hoặc theo dõi lâm sàng (2, 6, 11). Đối với VA trơ, verapamil hoặc propafenone cũng có thể có hiệu quả (9, 10, 12). Cấy ICD ở những bệnh nhân có nhịp nhanh thất đa hình do tiết catecholamine nên được dành riêng cho những bệnh nhân có SCA trước đó hoặc bệnh nhân có VAs trơ với điều trị thuốc phối hợp. Các cơn sốc không phù hợp được báo cáo ở 20% đến 30% bệnh nhân nhịp tim đa hình do tiết catecholamine với ICDs (2, 13-16). Việc lập trình ICD ở bệnh nhân nhịp nhanh thất đa hình do tiết catecholamine nên được tối ưu hóa để cung cấp liệu pháp cho VF và giảm thiểu các cú sốc không phù hợp và nguy cơ các cơn bão điện có thể gây tử vong (13, 15). Bóc bỏ giao cảm tim trái cho nhịp nhanh thất đa hình do tiết cathecholamine có thể giảm tần số sốc ICD tái phát bằng 32% xuống 75% (3-5, 17, 18) mặc dù ngất tái phát, SCA, hoặc SCD được báo cáo ở 9% đến 32% bệnh nhân, với các biến chứng nhỏ khác ở 20% đến 70% bệnh nhân. Tốt nhất nếu bóc vỏ giao cảm tim được thực hieenju ở ccs trung tâm với kinh nghiệm về thủ thuật này. Tăng cường điều trị y tế hoặc bóc giao cảm tim trái là quan trọng trong điều trị những người biểu hiện với sốc ICD phù hợp tái phát (19).
3. Test di truyền có thể hữu ích để xác định chẩn đoán nhịp nhanh thất đa hình do tiết catecholamine, được gợi ý bằng phát triển của VT hai chiều với gắng sức hoặc stress. Việc nhận biết nhịp nhanh thất đa hình do tiết catecholamine là nguyên nhân gây ra các triệu chứng gắng sức nên điều trị tích cực để ngăn ngừa nguy cơ SCD. Điều trị nhịp nhanh thất đa hình do tiết catecholamine không được hướng dẫn bằng trạng thái kiểu gene, nhưng việc kiểm tra những người thân thế hệ thứ nhất có thể làm dễ dàng với test di truyền (20). Các đột biến thụ thế ryanodine đã được báo cáo ở 47% số người tham gia thử nghiệm, đó là các đột biến de Novo > 70% (7). Trạng thái kiểu gene Ryanodine không tương quan với mức độ nghiêm trọng của bệnh hoặc đáp ứng với thuốc (7). Ở những bệnh nhân rất trẻ có VF nguyên phát, các đột biến ở caldulin đã được xác định và có liên quan đến tình trạng gây tử vong cao (21-24). Các nghiên cứu về các đột biến gây bệnh được đề xuất trong gen nhịp nhanh thất đa hình do tiết catecholamine cho thấy có đến 15% các biến thể đã có trong các cơ sở dữ liệu trước đây của quần thể nói chung, gây ra các câu hỏi về nguyên nhân đơn gene của nhịp nhanh thất đa hình do tiết catecholamine (20, 25).
Hình 13. VT đa hình tạo ra do gắng sức trong nhịp nhanh thất đa hình do tiết Catecholamine
Tài liệu tham khảo
1. Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426-34.
2. Roston TM, Vinocur JM, Maginot KR, et al. Catecholaminergic polymorphic ventricular tachycardia in children: analysis of therapeutic strategies and outcomes from an international multicenter registry. Circ Arrhythm Electrophysiol. 2015;8:633-42.
3. Collura CA, Johnson JN, Moir C, et al. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752-9.
4. Hofferberth SC, Cecchin F, Loberman D, et al. Left thoracoscopic sympathectomy for cardiac denervation in patients with life-threatening ventricular arrhythmias. J Thorac Cardiovasc Surg. 2014;147:404-9.
5. Schneider HE, Steinmetz M, Krause U, et al. Left cardiac sympathetic denervation for the management of life-threatening ventricular tachyarrhythmias in young patients with catecholaminergic polymorphic ventricular tachycardia and long QT syndrome. Clin Res Cardiol. 2013;102:33-42.
6. van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011;57:2244-54.
7. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
8. Leenhardt A, Lucet V, Denjoy I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995;91:1512-9.
9. Rosso R, Kalman JM, Rogowski O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149-54.
10.Swan H, Laitinen P, Kontula K, et al. Calcium channel antagonism reduces exercise-induced ventricular arrhythmias in catecholaminergic polymorphic ventricular tachycardia patients with RyR2 mutations. J Cardiovasc Electrophysiol. 2005;16:162-6.
11.Watanabe H, van der Werf C, Roses-Noguer F, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in kiểu gene-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542-7.
12.Hwang HS, Hasdemir C, Laver D, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2011;4:128-35.
13.Adler A, Sadek MM, Chan AY, et al. Patient outcomes from a specialized inherited arrhythmia clinic. Circ Arrhythm Electrophysiol. 2016;9:e003440.
14.Olde Nordkamp LR, Postema PG, Knops RE, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13:443-54.
15.Roses-Noguer F, Jarman JW, Clague JR, et al. Outcomes of defibrillator therapy in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2014;11:58-66.
16.Sy RW, Gollob MH, Klein GJ, et al. Arrhythmia characterization and long-term outcomes in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2011;8:864-71.
17.De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation. 2015;131:2185-93.
18.Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-9.
19.Zhang C, Kutyifa V, Moss AJ, et al. Long-QT syndrome and therapy for attention deficit/hyperactivity disorder. J Cardiovasc Electrophysiol. 2015;26:1039-44.
20.Jabbari J, Jabbari R, Nielsen MW, et al. New exome data question the pathogenicity of genetic variants previously associated with catecholaminergic polymorphic ventricular tachycardia. Circ Cardiovasc Genetics. 2013;6:481-9.
21.Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009-17.
22.Makita N, Yagihara N, Crotti L, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7:466-74.
23.Marsman RF, Barc J, Beekman L, et al. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J Am Coll Cardiol. 2014;63:259-66.
24.Nyegaard M, Overgaard MT, Sondergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703-12.
25.Paludan-Muller C, Ahlberg G, Ghouse J, et al. Integration of 60,000 exomes and ACMG guidelines question the role of catecholaminergic polymorphic ventricular tachycardia-associated variants. Clin Genet. 2017;91:63-72.
8. Rối loạn nhịp thất (VA) ở tim cấu trúc bình thường
|
Các khuyến cáo cho VA ở tim cấu trúc bình thường Tài liệu tham khảo ủng hộ các khuyến cáo được tóm tắt trong Tư liệu hỗ trợ online 45. |
||
|
COR |
LOE |
Khuyến cáo |
|
I |
B-R |
1. Ở các bệnh nhân có PVCs có triệu chứng ở tim bình thường trong tình trạng khác, điều trị bằng beta blocker hoặc chẹn canxi nondihydropyradine hữu ích để giảm loạn nhịp tái phát và cải thiện triệu chứng (1, 2). |
|
IIa |
B-R |
2. Ở các bệnh nhânVAcó triệu chứng ở tim bình thường ở tình trạng khác, điều trị bằng thuốc chống loạn nhịp là hợp lý để giảm các triệu chứng loạn nhịp tái phát và cải thiện triệu chứng nếu các thuốc chẹn beta và thuốc chẹn kênh canxi nondihydropyradine không có hiệu quả hoặc không dung nạp (3, 4). |
Tóm tắt
Hầu hết VA nguyên phát là do một cơ chế ổ của hoạt động khởi kích hoặc tính tự động bất thường, một số nhịp nhanh vào lại giữa các nhánh bó đã được ghi nhận. Các biểu hiện lâm sàng của VA nguyên phát rất biến đổi và dao động từ lành tính, PVCs không triệu chứng đến VT dai dẳng hoặc thậm chí VF. Trong phát hiện khởi đầu, đánh giá về bệnh tim cấu trúc được bảo đảm bằng khám thực thể, ECG và hình ảnh, thường là siêu âm tim. Trong trường hợp không có bất thường hoặc tiền sử gia đình có SCD, việc đánh giá và điều trị thêm sẽ được hướng dẫn bằng các triệu chứng. Nếu bệnh nhân không có triệu chứng và không có bằng chứng chứng về bệnh kênh tim, cần đảm bảo như đặc tính lành tự nhiên là đủ. Nếu loạn nhịp bị nghi ngờ thường xuyên liên tục gây ra rối loạn chức năng thất theo thời gian, nên theo dõi định kỳ để đánh giá lại chức năng thất (xem Phần 10.8). Đối với các triệu chứng nhẹ, tránh các yếu tố tăng nặng như sử dụng quá nhiều chất caffein hoặc các thuốc giống giao cảm, có thể là đủ. Liệu pháp với thuốc chẹn beta hoặc thuốc chẹn kênh canxi nondihydropyradine làm giảm triệu chứng cho một số bệnh nhân. Thuốc chống loạn nhịp nhóm I có thể có hiệu quả, nhưng thường tránh dùng vì các tác dụng phụ. Đối với những bệnh nhân đòi hỏi ức chế rối loạn nhịp, đối với các trường hợp này các thuốc chống rối loạn nhịp không hiệu quả, không dung nạp, hoặc không mong muốn, triệt phá qua catheter có thể là điều trị có hiệu quả cao (xem Phần 9). Chiến lược triệt phá để nhận biết vị trí nguồn gốc được biểu hiện bằng vị trí hoạt hóa điện học sớm nhất hoặc, khi điều này không khả thi, bằng lập bản đồ tạo nhịp (pace – mapping). Vị trí nguồn gốc thông thường nhất đối với VA nguyên phát là từ đường ra thất phải (RVOT) hoặc hoặc các lỗ nhỏ (ostium) của LV, bao gồm chỗ mở hình bầu dục của LV đến phần động mạch chủ được gắn vào phía trước và nhĩ trái được gắn vào phía sau. Nguồn gốc có thể có thể được dự đoán một cách hợp lý từ hình thái QRS của VA, điều này cung cấp chỉ dẫn tốt về loại phương pháp tiếp cận được đòi hỏi và khả năng thành công và nguy cơ. Triệt phá thất bại thường liên quan đến không có VA cho việc lập bản đồ khi thủ thuật, hoặc nguồn gôc VA ở khi vực của tim không tiếp cận được. Các ổ này đôi khi tạo ra VT đơn hình dai dẳng (5-7).
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
1. Trong một nghiên cứu đối chứng ngẫu nhiên, mù đôi, giả dược trên 52 bệnh nhân VA có triệu chứng và số lượng PVC trung bình 21.407 ± 1740 nhịp mỗi 24 giờ, atenolol làm giảm đáng kể tần số triệu chứng (p = 0.03) và số lượng PVC (p = 0,001), trong khi giả dược không ảnh hưởng đến số lượng PVC (p = 0,78) hoặc nhịp tim trung bình (p = 0,44) (8). Một so sánh ngẫu nhiên về thuốc chống loạn nhịp với triệt phá qua catheter, metoprolol hoặc propafenone có hiệu quả khiêm tốn để ức chế RVOT-VA mặc dù tỷ lệ tái phát cao hơn rất nhiều so với triệt phá qua catheter (9).
2. Trong một nghiên cứu RCT trên 233 bệnh nhân có ≥30 PVCs mỗi giờ, d-sotalol đã được chứng minh làm giảm lượng PVC thường xuyên, nhưng chỉ có racemic dl-sotalol hiện có sẵn (10). Trong một so sánh ngẫu nhiên ngẫu nhiên các thuốc chống loạn nhịp với triệt phá qua catheter, điều trị metoprolol hoặc propafenone có hiệu quả khi sử dụng để ức chế RVOT PVC mặc dù tỷ lệ tái phát cao hơn rất nhiều so với triệt phá qua catheter (9). Các thuốc chẹn kênh canxi Nydihydropyridine làm giảm loạn nhịp (1, 2, 11, 12).
Tài liệu tham khảo
1. Gill JS, Blaszyk K, Ward DE, et al. Verapamil for the suppression of idiopathic ventricular tachycardia of left bundle branch blốc-like morphology. Am Heart J. 1993;126:1126-33.
2. Gill JS, Ward DE, Camm AJ. Comparison of verapamil and diltiazem in the suppression of idiopathic ventricular tachycardia. Pacing Clin Electrophysiol. 1992;15:2122-6.
3. Kontos MC, Diercks DB, Ho PM, et al. Treatment and outcomes in patients with myocardial infarction treated with acute beta-blocker therapy: results from the American College of Cardiology’s NCDR(R). Am Heart J. 2011;161:864-70.
4. Levine JH, Massumi A, Scheinman MM, et al. Intravenous amiodarone for recurrent sustained hypotensive ventricular tachyarrhythmias. Intravenous Amiodarone Multicenter Trial Group. J Am Coll Cardiol. 1996;27:67-75.
5. Noda T, Shimizu W, Taguchi A, et al. Malignant entity of idiopathic ventricular fibrillation and polymorphic ventricular tachycardia initiated by premature extrasystoles originating from the right ventricular outflow tract. J Am Coll Cardiol. 2005;46:1288-94.
6. Viskin S. Idiopathic polymorphic ventricular tachycardia: a “benign disease” with a touch of bad luck? Korean Circ J. 2017;47:299-306.
7. Viskin S, Rosso R, Rogowski O, et al. The “short-coupled” variant of right ventricular outflow ventricular tachycardia: a not-so-benign form of benign ventricular tachycardia? J Cardiovasc Electrophysiol. 2005;16:912-6.
8. Krittayaphong R, Bhuripanyo K, Punlee K, et al. Effect of atenolol on symptomatic ventricular arrhythmia without structural heart disease: a randomized placebo-controlled study. Am Heart J. 2002;144:e10. efficacy when used to suppress RVOT PVCs although with a far higher rate of recurrence than catheter ablation (9). Nondihydropyridine calcium channel blockers reduce arrhythmias (1, 2, 11, 12).
9. Ling Z, Liu Z, Su L, et al. Radiofrequency ablation versus antiarrhythmic medication for treatment of ventricular premature beats from the right ventricular outflow tract: prospective randomized study. Circ Arrhythm Electrophysiol. 2014;7:237-43.
10.Hohnloser SH, Meinertz T, Stubbs P, et al. Efficacy and safety of d-sotalol, a pure class III antiarrhythmic compound, in patients with symptomatic complex ventricular ectopy. Results of a multicenter, randomized, double-blind, placebo-controlled dose-finding study. The d-Sotalol PVC Study Group. Circulation. 1995;92:1517-25.
11.Badhwar N, Scheinman MM. Idiopathic ventricular tachycardia: diagnosis and management. Curr Probl Cardiol. 2007;32:7-43.
12.Connolly SJ. Meta-analysis of antiarrhythmic drug trials. Am J Cardiol. 1999;84:90R-3R.
8.1. Rối loạn nhịp thất (VA) đường thoát và vòng nhĩ thất
|
Các khuyến cáo cho VA đường thoát Tài liệu tham khảo hỗ trợ các khuyến cáo được tóm tắt trong tư liệu hỗ trợ Online 46. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân VA đường thoát có triệu chứng ở tim bình thường khác đối với họ các thuốc chống loạn nhịp không hiệu quả, không dung nạp, hoặc bệnh nhân không ưa chuộng, triệt phá qua catheter là hữu ích (1-3). |
|
I |
B-NR |
2. Ở các bệnh nhân VT đường thoát có triệu chứng ở tim bình thường khác, beta blocker hoặc chẹn kênh canxi là hữu ích (1-3). |
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
1. Trong 1 RCT, việc triệt phá qua catheter ưu thế hơn các thuốc chống loạn nhịp trong việc ngăn chặn các PVC thường xuyên phát sinh từ RVOT (4). Các nghiên cứu quan sát đã cho thấy triệt phá qua catheter bằng năng lượng tần số radio có hiệu quả trong điều trị VA nguyên phát phát sinh từ RVOT và đường ra LV (2, 5-16). Vị trí triệt phát có thể là ở dưới hoặc ở trên van phổi trong RVOT (9, 13). Mặc dù hầu hết các RVOT VA có thể được triệt phá trong phạm vi RV, 10% có thể đòi hỏi triệt phá trong phạm vi đỉnh của đường cong (cusp) xoang phổi (9). Các biến chứng nghiêm trọng thường ít. Đối với VA đường ra LV, vị trí triệt phá có thể trong phạm vi xoang đỉnh đường cong (cusp) động mạch chủ (11, 14, 16), dưới van động mạch chủ (2, 6), ở khoảng nối tiếp hai lá động mạch chủ (1-3) hoặc trên bề mặt thượng tâm đỉnh LV (3, 17, 18). Vòng van hai lá và ba lá vị trí ít gặp của VA nguyên phát, nhưng VA này có thể cũng được điều trị có hiệu quả bằng triệt phá qua catheter (1, 19, 20). Khoảng 10% VA nguyên phát có thể phát sinh từ đỉnh của LV. Một số có thể được được triệt phá từ tĩnh mạch tim lớn hoặc bề mặt thượng tâm mạc, nhưng các vị trí khác phát sinh từ khi vực không thể tiếp cận ở sát với động mạch vành trái tháo dỡ khỏi tĩnh mạch tim hoặc trên bề mặt nhĩ, nhưng một số khác lại xuất hiện từ một vùng không thể tiếp cận gần động mạch vành trái, làm mất đi hiệu quả triệt phá (14). Các vị trí nguồn gốc trong cơ ít gặp nhưng có thể đòi hỏi triệt phá trên bề mặt cả hai nội tâm mạc và thượng tâm mạc của các lỗ LV (3). Các biến chứng do triệt phá VA đường thoát ít gặp, nhưng biến chứng chảy máu liên quan đến tiếp cận động mạch và tĩnh mạch, ép ngoại tâm mạc (tamponade), tổn thương đến các động mạch vành có thể xảy ra.
2. Trong một so sánh ngẫu nhiên tiền cứu các thuốc chống loạn nhịp đối lại với triệt phá qua catherter, metoprolol hoặc propafenone tỏ ra có hiệu quả khiêm tốn khi dùng để ức chế RVOT PVC, mặc dù tỷ lệ tái phát cao hơn rất nhiều so với triệt phá qua catheter (4). Các thuốc chẹn kênh canxi nondihydropyradine ngăn chặn loạn nhịp ở một số bệnh nhân (4).
Tài liệu tham khảo
1. Tada H, Ito S, Naito S, et al. Idiopathic ventricular arrhythmia arising from the mitral annulus: a distinct subgroup of idiopathic ventricular arrhythmias. J Am Coll Cardiol. 2005;45:877-86.
2. Yamada T, Litovsky SH, Kay GN. The left ventricular ostium: an anatomic concept relevant to idiopathic ventricular arrhythmias. Circ Arrhythm Electrophysiol. 2008;1:396-404.
3. Yamada -T, Maddox WR, McElderry HT, et al. Radiofrequency catheter ablation of idiopathic ventricular arrhythmias originating from intramural foci in the left ventricular outflow tract: efficacy of sequential versus simultaneous unipolar catheter ablation. Circ Arrhythm Electrophysiol. 2015;8:344-52.
4. Ling Z, Liu Z, Su L, et al. Radiofrequency ablation versus antiarrhythmic medication for treatment of ventricular premature beats from the right ventricular outflow tract: prospective randomized study. Circ Arrhythm Electrophysiol. 2014;7:237-43.
5. Carballeira Pol L, Deyell MW, Frankel DS, et al. Ventricular premature depolarization QRS duration as a new marker of risk for the development of ventricular premature depolarization-induced cardiomyopathy. Heart Rhythm. 2014;11:299-306.
6. Kamioka M, Mathew S, Lin T, et al. Electrophysiological and electrocardiographic predictors of ventricular arrhythmias originating from the left ventricular outflow tract within and below the coronary sinus cusps. Clin Res Cardiol. 2015;104:544-54.
7. Konstantinidou M, Koektuerk B, Wissner E, et al. Catheter ablation of right ventricular outflow tract tachycardia: a simplified remote-controlled approach. Europace. 2011;13:696-700.
8. Latchamsetty R, Yokokawa M, Morady F, et al. Multicenter outcomes for catheter ablation of idiopathic premature ventricular complexes. JACC Clin Electrophysiol. 2015;1:116-23.
9. Liao Z, Zhan X, Wu S, et al. Idiopathic ventricular arrhythmias originating from the pulmonary sinus cusp: prevalence, electrocardiographic/electrophysiological characteristics, and catheter ablation. J Am Coll Cardiol. 2015;66:2633-44.
10.Morady F, Kadish AH, DiCarlo L, et al. Long-term results of catheter ablation of idiopathic right ventricular tachycardia. Circulation. 1990;82:2093-9.
11.Ouyang F, Fotuhi P, Ho SY, et al. Repetitive monomorphic ventricular tachycardia originating from the aortic sinus cusp: electrocardiographic characterization for guiding catheter ablation. J Am Coll Cardiol. 2002;39:500-8.
12.Pedersen CT, Kay GN, Kalman J, et al. EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Heart Rhythm. 2014;11:e166-96.
13.Tada H, Tadokoro K, Miyaji K, et al. Idiopathic ventricular arrhythmias arising from the pulmonary artery: prevalence, characteristics, and topography of the arrhythmia origin. Heart Rhythm. 2008;5:419-26.
14.Yamada T, McElderry HT, Doppalapudi H, et al. Idiopathic ventricular arrhythmias originating from the aortic root prevalence, electrocardiographic and electrophysiologic characteristics, and results of radiofrequency catheter ablation. J Am Coll Cardiol. 2008;52:139-47.
15.Yamada T, McElderry HT, Doppalapudi H, et al. Idiopathic ventricular arrhythmias originating from the left ventricular summit: anatomic concepts relevant to ablation. Circ Arrhythm Electrophysiol. 2010;3:616-23.
16.Yamada T, McElderry HT, Okada T, et al. Idiopathic left ventricular arrhythmias originating adjacent to the left aortic sinus of valsalva: electrophysiological rationale for the surface electrocardiogram. J Cardiovasc Electrophysiol. 2010;21:170-6.
17.Mountantonakis SE, Frankel DS, Tschabrunn CM, et al. Ventricular arrhythmias from the coronary venous system: prevalence, mapping, and ablation. Heart Rhythm. 2015;12:1145-53.
18.Nagashima K, Choi EK, Lin KY, et al. Ventricular arrhythmias near the distal great cardiac vein: challenging arrhythmia for ablation. Circ Arrhythm Electrophysiol. 2014;7:906-12.
19.Hai JJ, Chahal AA, Friedman PA, et al. Electrophysiologic characteristics of ventricular arrhythmias arising from the aortic mitral continuity-potential role of the conduction system. J Cardiovasc Electrophysiol. 2015;26:158-63.
20.Tada H, Tadokoro K, Ito S, et al. Idiopathic ventricular arrhythmias originating from the tricuspid annulus: prevalence, electrocardiographic characteristics, and results of radiofrequency catheter ablation. Heart Rhythm. 2007;4:7-16.
8.2. VA cơ nhú (Papillary Muscle VA)
|
Khuyến cáo cho VA cơ nhú (PVCs và VT) Tài liệu tham khảo ủng hộ các khuyến cáo được tóm tắt trong tư liệu hỗ trợ online 47. |
||
|
COR |
LOE |
Khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân VA có triệu chứng phát sinh từ cơ nhú đối với những người các thuốc chống loạn nhịp không có hiệu quả, không dung nạp, hoặc không được bệnh nhân ưa thích, triệt phá qua catheter là hữu ích (1-5). |
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
1. Cơ nhú của LV hoặc RV có thể là vị trí nguồn gốc của VA có thể có hoặc không có bệnh tim cấu trúc (1-5). VA cơ nhú thất trái và thất phải nguyên phát thường nhất là PVCs và NSVT, và thường liên quan đến gắng sức và có thể tạo ra bằng truyền epinephrine hoặc isoproterenol tĩnh mạch (3). Các loạn nhịp này có cơ chế ổ, không phải vào lại. Bất kỳ cơ nhú RV (3) nào cũng có thể là vị trí nguồn gốc và triệt phá qua catheter thường cơ hiệu quả (2). Trong 1 nghiên cứu, triệt phá thành công đã đạt được ở tất cả 8 bệnh nhân với giảm gánh nặng PVC tử 17±20% đến 0.6±0.8% (2). Ở thất trái, vị trí nguồn gốc có thể hoặc cơ nhú sau giữa hoặc trước bên (1, 4, 5). Nhiều hình thái QRS của VA được quan sát ở 47% bệnh nhân, triệt phá ở cả hai mặt của cơ nhú cần thiết ở vài bệnh nhân (4). Đạt được ổn định catheter phù hợp có thể là một thách thức. Thành công cấp thời cao, nhưng tái phát nhiều hơn so với VA đường thoát nguyên phát. Các biến chứng nặng, gồm tổn thương van, thường ít gặp. Các nguy cơ triệt phá qua catheter gồm chảy máu liên quan đến tiếp cận động mạch và tĩnh mạch và nguy cơ thấp của tamponade màng ngoài tim.
Tài liệu tham khảo
1. Ban JE, Lee HS, Lee DI, et al. Electrophysiological characteristics related to outcome after catheter ablation of idiopathic ventricular arrhythmia originating from the papillary muscle in the left ventricle. Korean Circ J. 2013;43:811-8.
2. Crawford T, Mueller G, Good E, et al. Ventricular arrhythmias originating from papillary muscles in the right ventricle. Heart Rhythm. 2010;7:725-30.
3. Doppalapudi H, Yamada T, McElderry HT, et al. Ventricular tachycardia originating from the posterior papillary muscle in the left ventricle: a distinct clinical syndrome. Circ Arrhythm Electrophysiol. 2008;1:23-9.
4. Yamada T, Doppalapudi H, McElderry HT, et al. Electrocardiographic and electrophysiological characteristics in idiopathic ventricular arrhythmias originating from the papillary muscles in the left ventricle: relevance for catheter ablation. Circ Arrhythm Electrophysiol. 2010;3:324-31.
5. Yokokawa M, Good E, Desjardins B, et al. Predictors of successful catheter ablation of ventricular arrhythmias arising from the papillary muscles. Heart Rhythm. 2010;7:1654-9.
8.3. VT vào lại nhánh bó (nhịp nhanh Belhassen)
|
Khuyến cáo cho VT nhánh bó (nhịp nhanh Belhassen) Tài liệu tham khảo hỗ trợ các khuyến cáo được tóm tắt trong phần Bổ sung Dữ liệu online 48. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân nhạy cảm với verapamil, LVT nguyên phát liên quan đến vào lại nhánh bó đối với họ các thuốc chống loạn nhịp không hiệu quả, không dung nạp, hoặc bệnh nhân không ưa thích, triệt phá qua catheter là hữu ích (1-3). |
|
I |
B-NR |
2. Ở các bệnh nhân LVT nhạy cảm với verapamil huyết động ổn định lâu dài liên quan đến vào lại nhánh bó, tiêm tĩnh mạch verapamil để cắt cơn VT được khuyến cáo (3-6). |
|
IIa |
C-LD |
3. Ở các bệnh nhân LVT nguyên phát nhạy cảm với verapamil tái phát tái phát, điều trị lâu dài bằng uống verapamil có thể hữu ích (7-10). |
Văn bản Hỗ trợ Riêng biệt- Khuến cáo
1. Nhịp nhanh thất bên trái (LVT)nguyên phát do vào lại liên quan đến một phần của hệ thống LV Purkinje, thường bó nhánh sau trái khi nhánh dẫn truyền ngược của vòng và được xác định không hoàn toàn một phần tổ chức LV khi nhánh dẫn xuôi, phân chia của nó nhạy cảmvớiverapamil (1-3). Các VT này kéo dài một cách điển hình với QRS có cấu hình blốc nhánh bó phải với trục trên phải với một trục phía trên. Ít gặp hơn là trục VT phía dưới hoặc VT QRS tương đối hẹp xảy ra như là kết quả của các đường vào lại luân phiên, cũng liên quan một phần của hệ thống Purkinje. Thuốc chẹn beta hoặc verapamil thường cắt các cơn rối loạn nhịp này, nhưng thất bại để ngăn ngừa tái phát ở một số bệnh nhân(1-3). Mục tiêu triệt phá cho hình thái phổ biến nhất thường đưa vào đầu xa của nhánh xuôi của hệ thống Purkinje dọc theo phần dưới của váchthất trái(LV)gần chỗ nối của nó với bó sau trái. Triệt phá qua catheter thành công cấp thời ở > 90% bệnh nhân với nguy cơ tái phát khoảng 10%. VT này có thể giống với VA bó do cơ chế ổ ở các bó sau trái hoặc trước trái của hệ thống His-Purkinje LV. Các rối loạn nhịp bó này thường có cơ chế ổ với đích triệt phá qua catheter là vị trí hoạt động điện sớm nhất được ghi với điện thế bó trước tâm thu. Triệt phá qua catheter có hiệu quả cao đối với VA bó và liên bó. Các biến chứng nặng ít gặp và gồm chảy máu ở vị trí tiếp cận động mạch hoặc tĩnh mạch và nguy cơ nhỏ blốc nhánh bó hoặc blốc nhĩ thất.
2. LVT nguyên phát dựa trên cơ sở cơ chế vào lại liên quan đến tổ chức với các đặc tính dẫn truyền chậm dọc theo vách LV khi nhánh xuôi và bó sau trái bình thường của hệ His Purkinje như là nhánh ngược. Vùng dẫn truyền chậm nhậy cảmvớiverapamil (3-6). Các rối loạn nhịp này thường có hình thái blốc nhánh bó phải điển hình với trục trên, mặc dù sự đảo chiều của vòng có thể tạo ra QRS tương đối hẹp trong quá trình VT. Verapamil thường cắt cơn rối loạn nhịp này trong vùng dẫn truyền chậm xuôi (3-6).
3. Mặc dù không có báo cáo RCT đã được công bố nhưng việc sử dụng verapamil kéo dài cho VT nhạy cảm với verapamil đã được báo cáo để kiểm soát nhịp nhanh này ở nhiều bệnh nhân, gồm cả hai người lớn và trẻ em (5, 8-10).
Tài liệu tham khảo
1. Lin D, Hsia HH, Gerstenfeld EP, et al. Idiopathic fascicular left ventricular tachycardia: linear ablation lesion strategy for noninducible or nonsustained tachycardia. Heart Rhythm. 2005;2:934-9.
2. Liu Y, Fang Z, Yang B, et al. Catheter ablation of fascicular ventricular tachycardia: long-term clinical outcomes and mechanisms of recurrence. Circ Arrhythm Electrophysiol. 2015;8:1443-51.
3. Nogami A, Naito S, Tada H, et al. Demonstration of diastolic and presystolic Purkinje potentials as critical potentials in a macroreentry circuit of verapamil-sensitive idiopathic left ventricular tachycardia. J Am Coll Cardiol. 2000;36:811-23.
4. Belhassen B, Rotmensch HH, Laniado S. Response of recurrent sustained ventricular tachycardia to verapamil. Br Heart J. 1981;46:679-82.
5. German LD, Packer DL, Bardy GH, et al. Ventricular tachycardia induced by atrial stimulation in patients without symptomatic cardiac disease. Am J Cardiol. 1983;52:1202-7.
6. Tsuchiya T, Okumura K, Honda T, et al. Effects of verapamil and lidocaine on two components of the re-entry circuit of verapamil-senstitive idiopathic left ventricular tachycardia. J Am Coll Cardiol. 2001;37:1415-21.
7. Anderson JH, Tester DJ, Will ML, et al. Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet. 2016;9:259-65.
8. Ohe T, Shimomura K, Aihara N, et al. Idiopathic sustained left ventricular tachycardia: clinical and electrophysiologic characteristics. Circulation. 1988;77:560-8.
9. Snyder C, Bishara J, Darling R, et al. Verapamil-sensitive ventricular tachycardia in an infant. Congenit Heart Dis. 2006;1:124-6.
10.Wang JD, Fu YC, Jan SL, et al. Verapamil sensitive idiopathic ventricular tachycardia in an infant. Jpn Heart J. 2003;44:667-71.
8.4. VT đa hình / VF nguyên phát
|
Các khuyến cáo cho VT đa hình /VF nguyên phát Tài liệu hỗ trợ cho khuyến cáo được tóm tắt trong Tư liệu Hỗ trợ online 49. |
||
|
COR |
LOE |
Các khuyến cáo |
|
I |
B-NR |
1. Ở các bệnh nhân trẻ tuổi (<40 tuổi) với SCA không giải thích được, gần đuối nước không giải thích, hoặc ngất do gắng sức tái phát, những người không bị bệnh tim thiếu máu cục bộ hoặc các bệnh tim cấu trúc khác, nên đánh giá thêm về hội chứng loạn nhịp di truyền được khuyến cáo (1-8). |
|
I |
B-NR |
2. Ở các bệnh nhân được hồi sức từ SCA do VT đa hình hoặc VF, ICD được khuyến cáo nếu sống sót có ý nghĩa > 1 năm được khuyến cáo (9-13). |
|
I |
B-NR |
3. Đối với bệnh nhân có các cơn VF nguyên phát tái phát được khởi đầu bằng PVC có hình thái QRS đồng nhất, triệt phá qua catheter là hữu ích (11, 14). |
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
1. Khi kết hợp với đánh giá lâm sàng, xét nghiệm di truyền có thể chẩn đoán trong khoảng từ 13% đến 60% trẻ (< 40 tuổi) sống sót của SCA (3), với các kiểu gen phổ biến nhất được nhận biết liên quan với hội chứng QT dài, nhịp nhanh thất đa hình do tiết catecholamine, hội chứng Brugada (8). Các biến cố đuối nước / gần đuối nước đặc biệt liên quan với LQT1 và nhịp nhanh thất đa hình do tiết catecholamine; các đột biến di truyền trong hội chứng QT dài và nhịp nhanh thất đa hình do tiết catecholamine đã được nhận biết ở 23% bệnh nhân với các cơn gần đuối nước không giải thích được (15). Trong một nghiên cứu (6), ngừng tim liên quan đến gắng sức, đặc biệt ở trẻ em, có thể liên quan đến hội chứng QT dài, nhịp nhanh thất đa hình do tiết catecholamine, hoặc hội chứng QT dài qua trung gian calmodulin / triadin / các đột biến nhịp nhanh thất đa hình do tiết catecholamine, có thể đòi hỏi test di truyền chuyên biệt bổ xung (1, 2, 4, 16-18). Các tai nạn lái xe hơi đơn giản nên nỗ lực xem xét các nguyên nhân rối loạn nhịp. Hiệu suất xét nghiệm di truyền cao hơn nếu có tiền sử gia đình về SCD ở người trẻ. Việc giới thiệu đến các trung tâm xét nghiệm di truyền chuyên môn là rất quan trọng nếu chuyên môn địa phương không có.
2. VF khi không có các bệnh tim cấu trúc có thể nhận biết được hoặc các hội chứng rối loạn nhịp di truyền đã biết như nhịp thất trái đa hình, hội chứng QT dài, hội chứng QT ngắn, hội chứng Brugada, hoặc hội chứng sóng J thường là kết quả của các PVC khoảng ghép ngắn phát sinh từ hệ thống Purkinje ở hoặc tâm thất phải hay trái, hoặc ít phổ biến hơn, từ cơ tâm thất (9-13). Nguy cơ tái phát sau hồi sinh VF nguyên phát rất cao (12). Trong số 38 bệnh nhân liên tiếp từ 6 trung tâm khác nhau được triệt phá VF nguyên phát tiên phát khởi đầu bằng PVC khoảng ghép ngắn, 87% đã trải qua ≥ 2 cơn VF trong năm trước (12). Do VF nguyên phát có liên quan đến nguy cơ tái phát VF rất cao, ICD được chỉ định để ngăn ngừa SCD. Triệt phá qua catheter các ổ khởi kích đã chứng minh hiệu quả cao trong việc loại bỏ PVC lắp lại chúng tạo VF ở các bệnh nhân này (11). Trong thời gian theo dõi sau thủ thuật trung bình 63 tháng, có 7 bệnh nhân (18%) trong 38 bệnh nhân được thực hiện triệt phá qua catheter VF nguyên phát được tạo ra do các PVCs có khoảng ghép ngắn bị tái phát VF trong thời gian theo dõi trung bình 4 tháng. Năm trong số 7 bệnh nhân được triệt phá lặp đi lặp lại không có VF tái phát. Vì vậy, mặc dù việc triệt phá qua catheter rất hiệu quả trong VF nguyên phát, nguy cơ tái phát còn rất lớn sau thủ thuật thành công rõ ràng và bệnh nhân nên được bảo vệ với ICD. ICD dưới da có thể không phải là một liệu pháp tốt cho những bệnh nhân này do nguy cơ cao hơn của nhận cảm sóng T được thấy trong quần thể này; tuy nhiên, tư liệu còn hạn chế (10).
3. VF nguyên phát có thể được khởi đầu bằng PVC phát sinh từ các đường thoát hoặc hệ thống His-Purkinje trong phạm vi hoặc thất phải hoặc thất trái (11, 14, 19-21). Một số bệnh nhân có các chùm cơn VF (bão điện) biểu hiện điển hình như các PVCs khởi đầu VT đa hình / VF. PVC thường có hình thái QRS đồng nhất và khoảng ghép ngắn và có thể là đích cho triệt phá để kiểm soát rối loạn nhịp (11). Đối với PVCs từ hệ thống Purkinje, mục tiêu triệt phá là điện thế Purkinje tần số cao đi trước PVCs. Khi các cơn được tạo ra bằng các PVCs khoảng ghép ngắn phát triển từ các đường thoát, đích triệt phá là vị trí hoạt động thất sớm nhất. Các bệnh nhân VF nguyên phát hay có thời kỳ VT / VF thường xuyên xen kẽ với thời kỳ tương đối yên lặng (11, 14). Để tối đa hóa xác suấttriệt pháthành công, thủ thuật được thực hiện tốt nhất trong thời kỳ PVC thường xuyên. Các đợt không thường xuyên của VF có thể thích hợp với việc triệt phá nếu thường xuyên có PVC với các hình thái QRS đồng nhất. Khi PVCs có thể được xác định, triệt phá thành công cao, nhưng sự tái phát trễ xảy ra ở khoảng 10% bệnh nhân do đó việc cấy ICD là quan trọng ngay cả khi việc triệt phá thành công rất lớn. Những nguy cơ triệt phá qua catheter gồm chảy máu ở vị trí tiếp cận động mạch hoặc tĩnh mạch và nguy cơ nhỏdo chènép tim (tamponade). Việc điều trị bằng quinidine cấp thời và kéo dài có thể ngăn chặn tái phát các cơn VF ở một sốbệnh nhân(22).
Tài liệu tham khảo
1. Anderson JH, Tester DJ, Will ML, et al. Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet. 2016;9:259-65.
2. Dalal A, Czosek RJ, Kovach J, et al. Clinical presentation of pediatric patients at risk for sudden cardiac arrest. J Pediatr. 2016;
3. Kumar S, Peters S, Thompson T, et al. Familial cardiological and targeted genetic evaluation: low yield in sudden unexplained death and high yield in unexplained cardiac arrest syndromes. Heart Rhythm. 2013;10:1653-60.
4. Linzer M, Pritchett EL, Pontinen M, et al. Incremental diagnostic yield of loop electrocardiographic recorders in unexplained syncope. Am J Cardiol. 1990;66:214-9.
5. Solomon SD, Zelenkofske S, McMurray JJ, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352:2581-8.
6. Tester DJ, Medeiros-Domingo A, Will ML, et al. Unexplained drownings and the cardiac channelopathies: a molecular autopsy series. Mayo Clin Proc. 2011;86:941-7.
7. Tzimas I, Zingraf JC, Bajanowski T, et al. The role of known variants of KCNQ1, KCNH2, KCNE1, SCN5A, and NOS1AP in water-related deaths. Int J Legal Med. 2016;1575-9.
8. Wang D, Shah KR, Um SY, et al. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int. 2014;237:90-9.
9. Conte G, Caputo ML, Regoli F, et al. True idiopathic ventricular fibrillation in out-of-hospital cardiac arrest survivors in the Swiss Canton Ticino: prevalence, clinical features, and long-term follow-up. Europace. 2017;19:259-66.
10.Frommeyer G, Dechering DG, Kochhauser S, et al. Long-time “real-life” performance of the subcutaneous ICD in patients with electrical heart disease or idiopathic ventricular fibrillation. J Interv Card Electrophysiol. 2016;47:185-8.
11.Haïssaguerre M, Shoda M, Jais P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002;106:962-7.
12.Knecht S, Sacher F, Wright M, et al. Long-term follow-up of idiopathic ventricular fibrillation ablation: a multicenter study. J Am Coll Cardiol. 2009;54:522-8.
13.Leenhardt A, Glaser E, Burguera M, et al. Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation. 1994;89:206-15.
14.Haïssaguerre M, Shah DC, Jais P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet. 2002;359:677-8.
15.Albertella L, Crawford J, Skinner JR. Presentation and outcome of water-related events in children with long QT syndrome. Arch Dis Child. 2011;96:704-7.
16.Miyake CY, Motonaga KS, Fischer-Colbrie ME, et al. Risk of cardiac disease and observations on lack of potential predictors by clinical history among children presenting for cardiac evaluation of mid-exertional syncope. Cardiol Young. 2016;26:894-900.
17.Gula LJ, Klein GJ, Hellkamp AS, et al. Ejection fraction assessment and survival: an analysis of the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Am Heart J. 2008;156:1196-200.
18.Winkel BG, Yuan L, Olesen MS, et al. The role of the sodium current complex in a nonreferred nationwide cohort of sudden infant death syndrome. Heart Rhythm. 2015;12:1241-9.
19.Noda T, Shimizu W, Taguchi A, et al. Malignant entity of idiopathic ventricular fibrillation and polymorphic ventricular tachycardia initiated by premature extrasystoles originating from the right ventricular outflow tract. J Am Coll Cardiol. 2005;46:1288-94.
20.Sadek MM, Benhayon D, Sureddi R, et al. Idiopathic ventricular arrhythmias originating from the moderator band: electrocardiographic characteristics and treatment by catheter ablation. Heart Rhythm. 2015;12:67-75.
21.Van HH, Zado ES, Haqqani H, et al. Catheter ablation of ventricular fibrillation: importance of left ventricular outflow tract and papillary muscle triggers. Heart Rhythm. 2014;11:566-73.
22.Viskin S, Belhassen B. Idiopathic ventricular fibrillation. Am Heart J. 1990;120:661-71.
9. Bệnh cơ tim do PVC tạo ra (PVC-Induced Cardiomyopathy)
|
Các khuyến cáo cho bệnh cơ tim do PVC tạo ra Tài liệu tham khảo ủng hộ các khuyến cáo được tóm tắt trong tư liệu hỗ trợ online 50. |
||
|
COR |
LOE |
Các khuyến cao |
|
I |
B-NR |
1. Đối với những bệnh nhân cần chấn loại bỏ triệu chứng do rối loạn nhịp hoặc làm giảm chức năng thất nghi ngờ do PVCs thường xuyên (thường > 15% nhịp đập và chủ yếu là 1 hình thái học) và những người thuốc chống loạn nhịp không có hiệu quả, không dung nạp, hoặc bệnh nhân không ưa thích, triệt phá qua catheter là hữu ích (1, 2). |
|
IIa |
B-NR |
2. Ở các bệnh nhân với bệnh cơ tim do PVS tạo ra, điều trị bằng thuốc (ví dụ thuốc chẹn beta, amiodarone) là hợp lý để giảm nhịp tim tái phát và cải thiện triệu chứng và chức năng LV (3, 4). |
Văn bản Hỗ trợ Riêng biệt – Khuyến cáo
1. PVCs thường xuyên (thường> 15% toàn bộ số lượng các nhắt bóp) có thể tạo ra hình thái rối loạn chức năng LV có thể hồi phục (5-18). Tuy nhiên, đôi khi rất khó để xác định xem PVCs có gây rối loạn chức năng LV hay liệu rối loạn chức năng LV tiến triển có gây ra PVC thường xuyên hay không. Rối loạn chức năng LV có liên quan đến gánh nặng PVC lớn hơn (> 10% và thường> 20%), NSVT, sóng P ngược sau PVC, và PVC xen kẽ (interpolated) (6, 15). Trong một nghiên cứu tiền cứu về triệt phá qua catheter cho bệnh cơ tim do PVC gây ra, triệt phá thành công hoàn toàn ở 80% bệnh nhân (19). Chức năng LV bình thường trở lại trong vòng 6 tháng ở 82% trong số 22 bệnh nhân suy chức năng thất lúc đầu. Do đó, PVC thường xuyên là nguyên nhân có thể đảo ngược được của rối loạn chức năng LVvàcó thể được điều trị hiệu quả bằng triệt phá qua catheter. Rất khó để xác định xem liệu chức năng LV rõ ràng có phản ảnh chức năng LV suy hoặc không có khả năng để đánh giá chính xác chức năng thất trái do hoạt động ngoại vị thường xuyên. Ở những bệnh nhân có mật độ PVC cao với chức năng thất bình thường, điều trị tối ưu và giám sát để ngăn ngừa và phát hiện suy giảm chức năng thất.
2. Trong một nghiên cứu mù đôi với 30 bệnh nhân có hoặc không có bệnh tim thiếu máu cục bộ có > 30 PVCs mỗi giờ so sánh sotalol với propranolol, tác dụng thúc đẩy loạn nhịp hiện diện ở 1 bệnh nhân với sotalol. Không có sự khác biệt đáng kể về ức chế PVCs (sotalol 65%, propranolol 44%), với việc giảm trong ngoại tâm thu thất cặp (couplets) là 99% đối với sotalol và 49% đối với propranolol. Có sự gia tăng đáng kể về QTc ở bệnh nhân với sotalol (20). Trong một nghiên cứu đối chứng ngẫu nhiên, mù đôi với 674 bệnh nhân có HF và LVEF < 0.40 do thiếu máu cục bộ hoặc NICM và ≥10 PVCs mỗi giờ, amiodarone làm giảm đáng kể VA, nhịp tim chậm lại và có liên quan đến tăng LVEF 42% trong 2 năm với xu hướng giảm tử suất không có ý nghĩa thông kê (4). VA có bổ xung vào rối loạn chức năng thất ở các bệnh nhân này hay không còn chưa biết rõ.
Tài liệu tham khảo
1. Haïssaguerre M, Shah DC, Jais P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet. 2002;359:677-8.
2. Haïssaguerre M, Shoda M, Jais P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation. 2002;106:962-7.
3. Lee GK, Klarich KW, Grogan M, et al. Premature ventricular contraction-induced cardiomyopathy: a treatable condition. Circ Arrhythm Electrophysiol. 2012;5:229-36.
4. Singh SN, Fletcher RD, Fisher SG, et al. Amiodarone in patients with congestive heart failure and asymptomatic ventricular arrhythmia. Survival Trial of Antiarrhythmic Therapy in Congestive Heart Failure. N Engl J Med. 1995;333:77-82.
5. Baman TS, Lange DC, Ilg KJ, et al. Relationship between burden of premature ventricular complexes and left ventricular function. Heart Rhythm. 2010;7:865-9.
6. Ban JE, Park HC, Park JS, et al. Electrocardiographic and electrophysiological characteristics of premature ventricular complexes associated with left ventricular dysfunction in patients without structural heart disease. Europace. 2013;15:735-41.
7. Bogun F, Crawford T, Reich S, et al. Radiofrequency ablation of frequent, idiopathic premature ventricular complexes: comparison with a control group without intervention. Heart Rhythm. 2007;4:863-7.
8. Carballeira Pol L, Deyell MW, Frankel DS, et al. Ventricular premature depolarization QRS duration as a new marker of risk for the development of ventricular premature depolarization-induced cardiomyopathy. Heart Rhythm. 2014;11:299-306.
9. Del Carpio Munoz F, Syed FF, Noheria A, et al. Characteristics of premature ventricular complexes as correlates of reduced left ventricular systolic function: study of the burden, duration, coupling interval, morphology and site of origin of PVCs. J Cardiovasc Electrophysiol. 2011;22:791-8.
10.Deyell MW, Park KM, Han Y, et al. Predictors of recovery of left ventricular dysfunction after ablation of frequent ventricular premature depolarizations. Heart Rhythm. 2012;9:1465-72.
11.Hamon D, Blaye-Felice MS, Bradfield JS, et al. A new combined parameter to predict premature ventricular complexes induced cardiomyopathy: impact and recognition of epicardial origin. J Cardiovasc Electrophysiol. 2016;27:709-17.
12.Hasdemir C, Ulucan C, Yavuzgil O, et al. Tachycardia-induced cardiomyopathy in patients with idiopathic ventricular arrhythmias: the incidence, clinical and electrophysiologic characteristics, and the predictors. J Cardiovasc Electrophysiol. 2011;22:663-8.
13.Kanei Y, Friedman M, Ogawa N, et al. Frequent premature ventricular complexes originating from the right ventricular outflow tract are associated with left ventricular dysfunction. Ann Noninvasive Electrocardiol. 2008;13:81-5.
14.Kawamura M, Badhwar N, Vedantham V, et al. Coupling interval dispersion and body mass index are independent predictors of idiopathic premature ventricular complex-induced cardiomyopathy. J Cardiovasc Electrophysiol. 2014;25:756-2.
15.Niwano S, Wakisaka Y, Niwano H, et al. Prognostic significance of frequent premature ventricular contractions originating from the ventricular outflow tract in patients with normal left ventricular function. Heart. 2009;95:1230-7.
16.Olgun H, Yokokawa M, Baman T, et al. The role of interpolation in PVC-induced cardiomyopathy. Heart Rhythm. 2011;8:1046-9.
17.Yokokawa M, Good E, Crawford T, et al. Recovery from left ventricular dysfunction after ablation of frequent premature ventricular complexes. Heart Rhythm. 2013;10:172-5.
18.Zhong L, Lee YH, Huang XM, et al. Relative efficacy of catheter ablation vs antiarrhythmic drugs in treating premature ventricular contractions: a single-center retrospective study. Heart Rhythm. 2014;11:187-93.
19.Deyell MW, Park KM, Han Y, et al. Predictors of recovery of left ventricular dysfunction after ablation of frequent ventricular premature depolarizations. Heart Rhythm. 2012;9:1465-72.
20.Kubac G, Klinke WP, Grace M. Randomized double blind trial comparing sotalol and propranolol in chronic ventricular arrhythmia. Can J Cardiol. 1988;4:355-9.
10. VA và SCD liên quan đến các quần thể đặc biệt
10.1. Vận động viên thể thao
Ở vận động viên, phạm vi các VA từ PVCs đơn độc, cặp đôi, và NSVT, cho đến VT dai dẳng và SCA đưa đến SCA (1). Các PVCs và các đoạn NSVT lắp lại ngắn, đặc biệt khi không có bệnh tim cấu trúc, phổ biến hơn ở người không phải vận động viên, nhưng nhìn chung chúng lành tính, chỉ cần vận động hạn chế và hiếm khi dẫn tới việc bỏ thể thao (2, 3). Ngược lại, các đoạn NSVT dài hơn, đặc biệt xuất hiện khi gắng sức, VT daidẳng và SCA / SCD thường ít gặp, nhưng chúng có tỷ lệ cao hơn ở vận động viên so với các thông báo ở quần thể nói chung ở các nhóm lứa tuổi tương ứng. Các ước tính đã được báo cáo về SCD từ 1 / 53,703 vận động viên/năm trong cơ sở dữ liệu Hiệp hội Nghiệp đoàn Vận động viên Quốc gia (4) đến < 1 / 200,000 tại các học sinh trung học ở Minnesota (5). Trong số những nghiên cứu được đánh giá có các giao thức dịch tễ tốt hơn, các ước tính nằm trong khoảng 1 / 40.000 đến 1 / 80.000 (6). Những con số này so với nguy cơ chung về dân số là 1.0 đến 1.9 / 100.000 ở thanh thiếu niên và người lớn trẻ (7, 8). Hơn nữa, có vẻ như cả hai sự khác biệt giữa thể thao và giới tính trong mức độ nguy cơ, với nam giới có nguy cơ cao hơn nữ giới ở hầu hết các môn thể thao (7, 9), người da đen có nguy cơ cao hơn người da trắng, và các cầu thủ bóng rổ nam là nguy cơ cao nhất nhóm ở Hoa Kỳ, 1 / 5200 vận động viên-năm (4).
Một nghiên cứu gồm cả hai vận động viên cạnh tranh và giải trí cho thấy cả hai nhóm đều có nguy cơ cao hơn SCD so với dân số chung, với vận động viên giải trí có số lượng tích lũy cao hơn (7), SCD xảy ra ở lớn tuổi hơn và sự phân bố bệnh khác nhau. Dữ liệu về SCD sau khi tử vong ở các vận động viên cho thấy 25% đến 40% giải phẫu tử thi âm tính, cho thấy vai trò của rối loạn phân tử di truyền ở các nạn nhân này (4, 10, 11) và đối với các thành viên trong gia đình (12).
Một hạn chế khác của phân tích dữ liệu SCD ở vận động viên tập trung vào nguyên nhân không phải tim, một số trong đó có các biến cố giống tim. Các nguyên nhân không phải tim gồm rối loạn thần kinh cấp tính, các thuốc ma túy, cơn tấn công tim, tiêu cơ xương (rhabdomyolysis), rối loạn tế bào lưỡi liềm, tự sát và tai nạn (13, 14). Tuy nhiên, loạn nhịp ở vận động viên vẫn là nguyên nhân bệnh lý gây tử vong phổ biến nhất và nhiều trường hợp xảy ra như là biến cố tim đầu tiên.
Nguyên nhân cấu trúc phổ biến nhất của SCAs và SCDs trong các vận động viên ở Hoa Kỳ là bệnh cơ tim phì đại (HCM), tiếp theo là nguồn gốc bất thường của động mạch vành, với viêm cơ tim bổ xung một phần nhỏ hơn nhưng đáng kể (15). Ngoài ra, các chứng rối loạn di truyền khác cũng góp phần phân bố các nguyên nhân gây SCD ở vận động viên, nhiều người trong số đó có thể bị nghi ngờ hoặc xác định bằng tiền sử cẩn thận của gia đình và trước khi đánh giá ECG.
Nói chung, việc điều chỉnh rối loạn nhịp ở vận động viên tiếp theo là ở những người không phải vận động viên. Đối với các can thiệp, hiện nay thường có khuyến cáo sử dụngmáy khử rung ngoài tự động(AED)có khả năng huấn luyện và làm thuận lợi cho các vận động viên cạnh tranh (16), với những tuyên bố ít cụ thể hơn cho khả năng AED tại các tụ điểm về sự sẵn có của AED tại các địa điểm (ví dụ sân tennis) hoặc các tình huống (ví dụ như chạy bộ hoặc chạy nhóm nhỏ) ) trong đó vận động viên giải trí đang xảy ra.
Nhiều vận động viên đã có các thủ thuật sửa chữa (sửa chữa khiếm khuyết bẩm sinh hoặc phát triển như nguồn gốc bất thường của động mạch vành) (17, 18) đang được điều trị cho các rối loạn di truyền (19) hoặc đã cấy ICD (1) và có thể tham gia vận động viên tùy thuộc vào tính chất và mức độ nghiêm trọng của bệnh và với các biện pháp đề phòng và tư vấn thích hợp liên quan đến các nguy cơ tiềm ẩn cón lại (19, 20). Ví dụ, các vận động viên có các rối loạn mắc phải như viêm cơ tim được tư vấn chống lại gắng sức ít nhất 3 đến 6 tháng sau khi giải quyết bệnh.
Tài liệu tham khảo
1. Zipes DP, Link MS, Ackerman MJ, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 9: arrhythmias and conduction defects: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e315-25.
2. Biffi A, Pelliccia A, Verdile L, et al. Long-term clinical significance of frequent and complex ventricular tachyarrhythmias in trained athletes. J Am Coll Cardiol. 2002;40:446-52.
3. Verdile L, Maron BJ, Pelliccia A, et al. Clinical significance of exercise-induced ventricular tachyarrhythmias in trained athletes without cardiovascular abnormalities. Heart Rhythm. 2015;12:78-85.
4. Harmon KG, Asif IM, Maleszewski JJ, et al. Incidence, cause, and comparative frequency of sudden cardiac death in national collegiate athletic association athletes: a decade in review. Circulation. 2015;132:10-9.
5. Maron BJ, Haas TS, Ahluwalia A, et al. Incidence of cardiovascular sudden deaths in Minnesota high school athletes. Heart Rhythm. 2013;10:374-77.
6. Harmon KG, Drezner JA, Wilson MG, et al. Incidence of sudden cardiac death in athletes: a state-of-the-art review. Br J Sports Med. 2014;48:1185-92.
7. Marijon E, Tafflet M, Celermajer DS, et al. Sports-related sudden death in the general population. Circulation. 2011;124:672-81.
8. Meyer L, Stubbs B, Fahrenbruch C, et al. Incidence, causes, and survival trends from cardiovascular-related sudden cardiac arrest in children and young adults 0 to 35 years of age: a 30-year review. Circulation. 2012;126:1363-72.
9. Marijon E, Bougouin W, Perier MC, et al. Incidence of sports-related sudden death in France by specific sports and sex. JAMA. 2013;310:642-3.
10.Anderson JH, Tester DJ, Will ML, et al. Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet. 2016;9:259-65.
11.Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374:2441-52.
12.Semsarian C, Ingles J, Wilde AA. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J. 2015;36:1290-6.
13.Maron BJ, Haas TS, Ahluwalia A, et al. Demographics and epidemiology of sudden deaths in young competitive athletes: from the United States national registry. Am J Med. 2016;129:1170-7.
14.Yankelson L, Sadeh B, Gershovitz L, et al. Life-threatening events during endurance sports: is heat stroke more prevalent than arrhythmic death? J Am Coll Cardiol. 2014;64:463-9.
15.Maron BJ, Haas TS, Murphy CJ, et al. Incidence and causes of sudden death in U.S. college athletes. J Am Coll Cardiol. 2014;63:1636-43.
16.Link MS, Myerburg RJ, Estes NA 3rd. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 12: emergency action plans, resuscitation, cardiopulmonary resuscitation, and automated external defibrillators: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e334-8.
17.Brothers JA, Frommelt MA, Jaquiss RDB, et al. Expert consensus guideline: anomalous aortic origin of a coronary artery – American Association for Thoracic Surgery Clinical Practice Guidelines. J Thorac Cardiovasc Surg. 2017;153:1440-57.
18.Van Hare GF, Ackerman MJ, Evangelista JA, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 4: congenital heart disease: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e281-91.
19.Ackerman MJ, Zipes DP, Kovacs RJ, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 10: the cardiac channelopathies: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e326-9.
20.Maron BJ, Udelson JE, Bonow RO, et al. Eligibility and disqualification recommendations for competitive athletes with cardiovascular abnormalities: Task Force 3: Hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy and other cardiomyopathies, and myocarditis: a scientific statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132:e273-80.


{kind=link}