

Trưởng ban: PGS.TS PHẠM NGUYỄN VINH
Đồng trưởng ban: TS.BS NGUYỄN THỊ THU HOÀI
(…)
5. Đánh giá nguy cơ và dự phòng đột tử do tim
5.1 Đánh giá nguy cơ đột tử do tim
| Loại | MCC | Khuyến cáo về đánh giá nguy cơ đột tử do tim |
| 1 | B-NR | 1. Ở những bệnh nhân BCTPĐ, việc đánh giá nguy cơ đột tử do tim bằng biện pháp không xâm nhập một cách toàn diện và có hệ thống ở lần thăm khám đầu tiên và mỗi 1 đến 2 năm sau đó được khuyến cáo và nên bao gồm việc đánh giá các yếu tố nguy cơ sau(65–89) (Hình 1, Hình 3 và Bảng 4):
a. Tiền sử bệnh nhân có ngừng tim hoặc rối loạn nhịp thất kéo dài b. Tiền sử bệnh nhân có ngất mà trên lâm sàng nghi ngờ là do rối loạn nhịp c. Tiền sử gia đình có người thân đột tử do BCTPĐ khi còn trẻ, ngừng tim hoặc rối loạn nhịp thất kéo dài d. Độ dầy thành thất trái, phân suất tống máu và tình trạng phình mỏm thất trái e. Các cơn nhịp nhanh thất không kéo dài (NSVT) trên máy theo dõi điện tâm đồ liên tục lưu động. |
| 1 | B-NR | 2. Ở những bệnh nhân BCTPĐ được xác định là không có nguy cơ đột tử do tim cao hoặc những bệnh nhân mà quyết định đặt ICD còn chưa chắc chắn sau khi đã đánh giá lâm sàng bao gồm tiền sử bản thân / gia đình, siêu âm tim và máy theo dõi điện tâm đồ lưu động, hình ảnh cộng hưởng từ tim là hữu ích trong việc đánh giá độ dầy thành thất trái, phân suất tống máu, tình trạng phình mỏm thất trái và mức độ sợi hóa cơ tim bằng LGE(21,65,74,79–82,84,90) (Bảng 4). |
| 2a | B-NR | 3. Ở những bệnh nhân BCTPĐ từ 16 tuổi trở lên, việc đánh giá đường kính nhĩ trái và chênh áp tối đa qua đường ra thất trái trên siêu âm tim là hợp lý để hỗ trợ việc tính toán nguy cơ đột tử 5 năm dự đoán , từ đó đưa ra quyết định cấy ICD(66,86) (Bảng 4). |
Nội dung khuyến cáo
BCTPĐ đã được coi là nguyên nhân phổ biến nhất của đột tử do tim ở người trẻ Bắc Mỹ, một biến chứng rất nguy hiểm và rất dễ thấy được của bệnh lý tim do di truyền này(65). Trong số những bệnh nhân BCTPĐ, những bệnh nhân trẻ hơn có nguy cơ đột tử do tim cao hơn so với các bệnh nhân cao tuổi hơn(91). Tỷ lệ tích lũy 5 năm số biến cố đột tử do tim ở trẻ em bị BCTPĐ từ khi được chẩn đoán là 8% đến 10% biến cố đột tử ở thời kì thơ ấu(92,93). Dường như không có sự khác biệt giữa các giới và các chủng tộc về nguy cơ đột tử do tim(94).
Qua nhiều thập kỷ, nhiều nghiên cứu đã tập trung vào việc xác định các dấu ấn nguy cơ lâm sàng chính giúp phân tầng bệnh nhân theo mức độ nguy cơ để xác định nhóm bệnh nhân nguy cơ cao có thể là ứng viên cho việc dự phòng đột tử do tim bằng ICD(95). Nhờ chiến lược phân tầng nguy cơ và sự có mặt của ICD trong thực hành lâm sàng đã làm giảm đáng kể tỷ lệ tử vong do bệnh lý này(96,97). Một thang điểm dự đoán nguy cơ cũng được tạo ra để tính nguy cơ đột tử 5 năm dự đoán của bệnh nhân, giúp bổ sung cho việc phần tầng nguy cơ và đưa ra quyết định cấy ICD ở bệnh nhân trưởng thành(66,86). Sự phát triển của việc đánh giá nguy cơ đột tử do tim, bao gồm cả việc có thêm các dấu ấn nguy cơ mới, đã dẫn tới việc loại bỏ đáp ứng huyết áp bất thường với gắng sức trong đánh giá thường quy nguy cơ đột tử do tim.
Các dấu ấn nguy cơ đột tử do tim không xâm nhập thường sử dụng hiện nay (Bảng 4) được dùng để ước tính mức độ nguy cơ ở từng bệnh nhân BCTPĐ và để xác định những bệnh nhân nhận được lợi ích từ việc dự phòng nguyên phát bằng liệu pháp ICD(65), dựa vào tiền sử bệnh nhân và gia đình(70), các phương pháp không xâm nhập như siêu âm tim(73), theo dõi điện tâm đồ lưu động(78) và hình ảnh cộng hưởng từ tim(84). Vì nguy cơ đột tử do tim kéo dài trong nhiều thập kỉ, tái đánh giá nguy cơ đột tử định kì là một phần quan trọng trong việc đánh giá xuyên suốt ở hầu hết các bệnh nhân BCTPĐ(65).
Cân nhắc việc phân tầng nguy cơ ở những bệnh nhân trẻ em
Trước đây, việc phân tầng nguy cơ đột tử do tim ở trẻ em được dựa theo các dấu ấn nguy cơ từ các nghiên cứu ở bệnh nhân BCTPĐ người lớn. Nhiều nghiên cứu gợi ý rằng các yếu tố nguy cơ có khả năng hạn chế trong việc dự đoán đột tử do tim ở những bệnh nhân trẻ em(92). Các nghiên cứu gần đây gợi ý rằng một số yếu tố nguy cơ trên bệnh nhân người lớn có vai trò quan trọng ở những bệnh nhân trẻ em BCTPĐ(92,98). Các mô hình dự đoán nguy cơ cho trẻ em mắc BCTPĐ đã được phát triển nhưng vẫn chưa được sử dụng rộng rãi trong thực hành lâm sàng(93). Các yếu tố nguy cơ được đưa ra trong hướng dẫn này vẫn dựa trên những yếu tố nguy cơ của bệnh nhân trưởng thành và những thông tin sẵn có riêng cho bệnh nhân trẻ em(99,100). Tóm lại, việc đưa ra quyết định đặt ICD cần dựa trên đánh giá cá thể trên từng bệnh nhân, tính đến tất cả các dấu ấn nguy cơ theo từng độ tuổi, mức độ của các yếu tố nguy cơ đã được xác định, hồ sơ lâm sàng tổng thể, mức độ nguy cơ có thể chấp nhận đối với bệnh nhân và gia đình và nguy cơ biến chứng liên quan tới thiết bị cấy ghép, bao gồm tác động về tâm lý và máy ICD sốc không thích hợp.
- Qua nhiều thập kỉ, nhiều nghiên cứu hồi cứu quan sát ở những bệnh nhân BCTPĐ đã xác định các thành phần trong tiền sử bản thân và gia đình cũng như các kết quả từ hình ảnh học tim mạch và theo dõi điện tâm đồ lưu động có liên quan tới gia tăng nguy cơ rối loạn nhịp thất đe dọa tính mạng trong tương lai(65–86). Do đó, đánh giá nguy cơ đột tử do tim ở lần thăm khám đầu tiên và tái đánh giá mỗi 1 đến 2 năm là một phần quan trọng trong đánh giá những bệnh nhân BCTPĐ và cần bao gồm: 1) tiền sử ngừng tim hoặc rối loạn nhịp thất kéo dài (trên 30 giây hoặc có kèm rối loạn huyết động); 2) tiền sử gia đình có người đột tử, ngừng tim hoặc rối loạn nhịp thất kéo dài được xác định là do hoặc nhiều khả năng do BCTPĐ ở ít nhất 1 thành viên có quan hệ huyết thống bậc 1 hoặc các thành viên ruột thịt trong gia đình ở tuổi ≤ 50; 3) theo dõi điện tâm đồ lưu động liên tục (24 đến 48 giờ) để phát hiện cơn nhanh thất kéo dài hoặc không kéo dài; 4) tiền sử gần đây có ngất (mất ý thức thoáng qua) khả năng là do rối loạn nhịp (những cơn xảy ra trong vòng 6 tháng vì nó có vai trò quan trọng nhất việc tiên lượng, trong khi những cơn trên 5 năm trở về trước có ít vai trò hơn); và 5) hình ảnh học tim mạch giúp xác định kích thước thành thất trái tối đa ở tất cả các thành của buồng thất trái(71,73), phân suất tống máu(88) và sự có mặt của phình mỏm(75,76). Ở những bệnh nhân trẻ em, kích thước thành thất trái thường được trả kết quả bằng giá trị tuyệt đối và giá trị hiệu chỉnh Z-score tiêu chuẩn theo diện tích bề mặt cơ thể. Do các dữ liệu gợi ý tỷ lệ biến cố đột tử do tim thấp hơn ở những bệnh nhân BCTPĐ cao tuổi (trên 60 tuổi), ổn định(97), việc quyết định đánh giá nguy cơ tiếp theo là cá thể hóa ở nhóm bệnh nhân này.
- So với hình ảnh cộng hưởng từ tim, siêu âm tim có thể đánh giá thấp hơn bề dầy tối đa thành thất trái và có thể không phát hiện ra phình mỏm ở một số bệnh nhân BCTPĐ(81). Hơn nữa, sự xơ hoá cơ tim lan rộng được phát hiện trên cộng hưởng từ tim, có liên quan tới gia tăng nguy cơ rối loạn nhịp thất đe dọa tính mạng(84). Do những lý do này, nếu một bệnh nhân BCTPĐ không có bằng chứng của tăng nguy cơ đột tử do tim sau khi đánh giá bằng tiền sử bản thân / gia đình, siêu âm tim và theo dõi điện tâm đồ lưu động, hoặc việc phân tầng nguy cơ chưa chắc chắn, hình ảnh cộng hưởng từ tim có chất tương phản có thể cung cấp thêm các đặc điểm về kích thước thành thất trái, phân suất tống máu, sự có mặt của phình mỏm thất trái và sự có mặt / mở rộng của vùng tăng tín hiệu Gadolinium muộn. Mặc dù hình ảnh cộng hưởng từ tim có thể có ích ở những bệnh nhân BCTPĐ trẻ em, nó có thể cần thuốc an thần và nguy cơ khi dùng thuốc có thể cao hơn so với lợi ích ở những trẻ em không có triệu chứng. Việc sử dụng hình ảnh cộng hưởng từ tim nên được quyết định bởi bác sĩ lâm sàng và gia đình sau khi đánh giá nguy cơ của từng trẻ em.
- Để tính toán thang điểm nguy cơ đột tử do tim 5 năm ở những bệnh nhân người lớn BCTPĐ, cần siêu âm tim đánh giá đường kính nhĩ trái và chênh áp tức thời tối đa qua đường ra thất trái bằng phương pháp Doppler liên tục. Việc ước tính thang điểm nguy cơ đột tử do tim không tính tới sự ảnh hưởng của các dấu ấn nguy cơ đột tử do tim mới như rối loạn chức năng tâm thu thất trái (PSTM dưới 50%), phình mỏm và vùng tăng tín hiệu Gadolinium muộn. Sự tác động của trên một dấu ấn nguy cơ này ở những bệnh nhân BCTPĐ đối với việc ước tính nguy cơ 5 năm là chưa thể xác định được.
5.2 Lựa chọn bệnh nhân cấy ICD
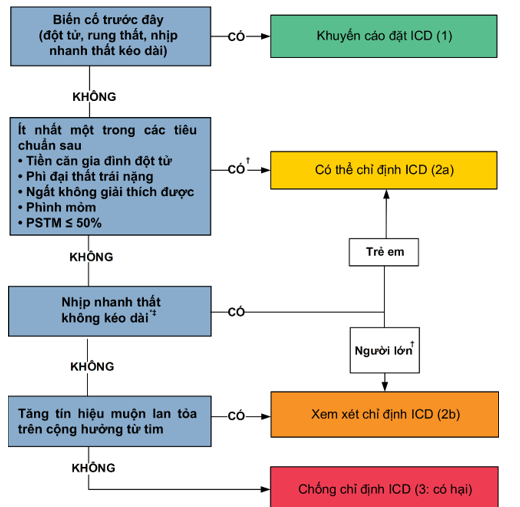
Hình 3. Chọn lọc bệnh nhân đặt máy phá rung cấy được (26)
* Các quyết định ICD ở bệnh nhi BCTPĐ dựa trên ≥ 1 trong số các yếu tố nguy cơ chính: tiền sử gia đình đột tử do BCTPĐ, nhịp nhanh thất không kéo dài trên màn hình theo dõi lúc cấp cứu, dầy thất trái nặng và ngất không rõ nguyên nhân.
† Ở bệnh nhân trên 16 tuổi, ước tính nguy cơ 5 năm có thể được xem xét để cung cấp thông tin đầy đủ cho bệnh nhân trong quá trình thảo luận chung ra quyết định.
‡ Có vẻ thích hợp nhất nếu xem xét nhịp nhanh thất xuất hiện thường xuyên, kéo dài và dễ xuất hiện cơn.
ICD, Implantable Cardioverter Defibrillator, Máy phá rung cấy được
| Loại | MCC | Khuyến cáo về cấy ICD ở những bệnh nhân BCTPĐ nguy cơ cao |
| 1 | C-EO | 1. Ở những bệnh nhân BCTPĐ, sử dụng đánh giá lâm sàng cá thể hóa được khuyến cáo khi đánh giá sức mạnh tiên lượng của các dấu ấn nguy cơ thông thường trong hồ sơ lâm sàng của từng bệnh nhân, cũng như thảo luận kỹ lưỡng và cân bằng về bằng chứng, lợi ích và rủi ro ước tính thu hút sự tham gia tích cực của bệnh nhân được thông tin đầy đủ vào quá trình đưa ra quyết định cấy máy phá rung cấy được (ICD). |
| 1 | B-NR | 2. Ở những bệnh nhân BCTPĐ và có tiền sử ngừng tim hoặc nhịp nhanh thất kéo dài được ghi nhận trong hồ sơ, cấy ICD được khuyến cáo (Hình 3, Bảng 4). |
| 2a | B-NR | 3. Ở những bệnh nhân BCTPĐ trưởng thành với ≥ 1 yếu tố nguy cơ chính với đột tử do tim, cấy ICD là hợp lý. Các yếu tố nguy cơ chính bao gồm (Hình 3, Bảng 4):
a. Đột tử được đánh giá là chắc chắn hoặc có vẻ do BCTPĐ ở ít nhất 1 thành viên có quan hệ huyết thống bậc 1 hoặc các thành viên ruột thịt trong gia đình ở tuổi ≤ 50; b. Thất trái phì đại rất dầy ≥ 30 mm ở bất kì vùng cơ tim thất trái; c. ≥ 1 cơn ngất gần đây nghi ngờ là do rối loạn nhịp dựa trên tiền sử lâm sàng (tức là không giống cơn ngất do thần kinh tim, hoặc do tắc nghẽn đường ra thất trái. d. Phình mỏm thất trái, không phụ thuộc kích thước; e. Rối loạn chức năng tâm thu thất trái (PSTM dưới 50%). |
| 2a | B-NR | 4. Ở những trẻ em BCTPĐ có ≥ 1 yếu tố nguy cơ thông thường, bao gồm ngất chưa rõ nguyên nhân, thất trái rất dầy, nhịp nhanh thất không kéo dài hoặc tiền sử gia đình có người đột tử do tim khi còn trẻ do BCTPĐ, cấy ICD là hợp lý sau khi cân nhắc tỷ lệ biến chứng dài hạn tương đối cao khi cấy ICD ở những bệnh nhân trẻ (Hình 3, Bảng 4). |
| 2a | B-NR | 5. Ở những bệnh nhân BCTPĐ ≥ 16 tuổi và có ≥ 1 yếu tố nguy cơ đột tử do tim chính, việc thảo luận về ước tính nguy cơ đột tử 5 năm và tỷ lệ tử vong có thể có ích trong quá trình đưa ra quyết định chung về việc cấy ICD (Hình 3, Bảng 4)(86,101). |
| 2b | B-NR | 6. Ở những bệnh nhân người lớn BCTPĐ và không có yếu tố nguy cơ đột tử do tim chính sau khi đánh giá lâm sàng, hoặc ở những bệnh nhân mà việc đưa ra quyết định cấy ICD chưa chắc chắn, ICD có thể được cân nhắc ở những bệnh nhân có vùng tăng tín hiệu muộn với Gadolinium trên hình ảnh cộng hưởng từ tim có chất tương phản hoặc xuất hiện nhịp nhanh thất không kéo dài trên kết quả theo dõi điện tâm đồ lưu động (Hình 3, Bảng 4)(65,101). |
| 2b | C-LD | 7. Ở những bệnh nhân trẻ em BCTPĐ mà việc phân tầng nguy cơ không chắc chắn, có thể hữu ích khi cân nhắc thêm các yếu tố như vùng tăng tín hiệu muộn Gadolinium lan rộng trên hình ảnh cộng hưởng từ tim có chất tương phản và rối loạn chức năng tâm thu trong việc phân tầng nguy cơ (Hình 3, Bảng 4). |
| 3: có hại | B-NR | 8. Ở những bệnh nhân BCTPĐ không có yếu tố nguy cơ, không nên cấy ICD. |
| 3: có hại | B-NR | 9. Ở những bệnh nhân BCTPĐ, không nên cấy ICD với mục đích duy nhất là tham gia các môn điền kinh. |
Nội dung khuyến cáo
Ở những bệnh nhân BCTPĐ, phân tầng nguy cơ và lựa chọn những bệnh nhân cho liệu pháp ICD dự phòng tiếp tục phát triển, bao gồm các dấu ấn nguy cơ mới và các chiến lược ước tính theo điểm. Hiệu quả đã được chứng minh của ICD trong việc điều trị các rối loạn nhịp nhanh thất đe dọa tính mạng và cứu sống những bệnh nhân BCTPĐ đã đặt ra tầm quan trọng ngày càng tăng của việc lựa chọn chính xác những bệnh nhân cho liệu pháp sử dụng thiết bị này(96,99). Qua nhiều thập kỉ, các nghiên cứu quan sát hồi cứu đã xác định một số các dấu ấn nguy cơ không xâm nhập trên lâm sàng có liên quan tới gia tăng nguy cơ biến cố đột tử trong BCTPĐ. Đi cùng với đánh giá lâm sàng và việc thảo luận để đưa ra quyết định, những bệnh nhân BCTPĐ được coi là ứng viên tiềm năng cho việc dự phòng nguyên phát bằng ICD khi có ≥ 1 dấu ấn nguy cơ chính, có độ nhạy cao trong việc dự đoán những bệnh nhân BCTPĐ có nguy cơ biến cố đột tử do tim cao nhất trong tương lai(65).
Gần đây đã xuất hiện các phương pháp tiếp cận khác để phân tầng nguy cơ trong BCTPĐ. Bằng việc kết hợp một số đặc điểm liên quan tới bệnh vào một phương trình hồi quy logistic, có thể ước tính nguy cơ đột tử trong 5 năm(92). Điểm nguy cơ trong BCTPĐ này có thể giúp những bệnh nhân hiểu được một ước tính định lượng về nguy cơ đột tử do tim của họ và được dùng trong việc thảo luận đưa ra quyết định. Bởi vì từng bệnh nhân có thể cân nhắc tác động của việc ước tính nguy cơ đột tử do tim khác nhau, chúng tôi thống nhất rằng không nên áp đặt các khuyến cáo điều trị đã được xác định trước cho các ước tính nguy cơ như là yếu tố duy nhất cho việc quyết định cấy ICD. Các dấu ấn nguy cơ đột tử do tim trong BCTPĐ bao gồm phình mỏm thất trái, LGE và rối loạn chức năng tâm thu (PSTM dưới 50%) không được đưa vào việc tính toán nguy cơ và tác động của chúng với thang điểm ước tính nguy cơ 5 năm là không chắc chắn.
- Việc ra quyết định cấy ICD dự phòng nguyên phát trong BCTPĐ thường phức tạp và là một thách thức, bởi vì tỷ lệ biến cố đột tử do tim quan sát được thấp ở bệnh lý này. Ngoài ra, những bệnh nhân BCTPĐ tương đối trẻ tuổi cần cân nhắc dự phòng đột tử do tim có nghĩa là thời gian nguy cơ thường có thể kéo dài trong nhiều năm và nhiều thập kỉ đối với mỗi bệnh nhân. Do những lý do này, việc quyết định liệu pháp ICD dự phòng nguyên phát nên kết hợp với việc thảo luận với bệnh nhân về nguy cơ đột tử do tim và lợi ích của liệu pháp ICD đã được chứng minh trong việc bảo vệ trước các rối loạn nhịp nhanh thất đe dọa tính mạng cân bằng với việc bệnh nhân hiểu được việc đặt máy kéo dài có thể đi kèm các biến chứng(102).
- Những bệnh nhân BCTPĐ đã trải qua ngừng tim hoặc nhịp nhanh thất / rung thất kèm rối loạn huyết động đáng kể vẫn có sự tăng nguy cơ đáng kể với rối loạn nhịp nhanh thất đe dọa tính mạng trong tương lai và do đó nên được cân nhắc liệu pháp ICD dự phòng thứ phát(102,103).
- Xác định những bệnh nhân BCTPĐ trưởng thành có nguy cơ đột tử do tim cao nên được hướng dẫn bằng sự có mặt của các yếu tố nguy cơ chính đột tử do tim không xâm nhập đã biết (Bảng 4). Bởi vì mỗi yếu tố nguy cơ chính này đều liên quan tới gia tăng nguy cơ, sẽ là hợp lý khi xem xét liệu pháp ICD nguyên phát ở những bệnh nhân có ≥ 1 yếu tố nguy cơ đột tử do tim (Hình 3 và Bảng 4). Chiến lược phân tầng nguy cơ này cung cấp độ nhạy cao trong việc xác định những bệnh nhân có thể nhận lợi ích từ liệu pháp ICD và là cơ hội để kết hợp đầy đủ quy trình đưa ra quyết định chung bao gồm việc xem xét hồ sơ lâm sàng đầy đủ của bệnh nhân cũng như đánh giá của bác sĩ và nguyện vọng của bệnh nhân. Tỷ lệ biến cố đột tử do tim quan sát được rất thấp ở những bệnh nhân BCTPĐ cao tuổi (trên 60 tuổi), chiến lược phân tầng nguy cơ với các dấu ấn chính thường được sử dụng ở những bệnh nhân BCTPĐ trưởng thành trẻ tuổi hoặc trung niên.
- Việc phân tầng nguy cơ ở trẻ em BCTPĐ cần đánh giá các yếu tố nguy cơ theo tuổi. Mặc dù ngất chưa rõ nguyên nhân, nhịp nhanh thất không kéo dài, kích thước thành thất trái và Z-score của đường kính nhĩ trái có tương quan với nguy cơ đột tử do tim ở bệnh nhân trẻ em tương tự như bệnh nhân trưởng thành (Bảng 4), mối tương quan giữa tuổi, chênh áp qua đường ra thất trái và tiền sử gia đình có người đột tử do tim có sự khác biệt với người trưởng thành(92). Trên cơ sở toàn bộ dữ liệu đã có và ý kiến chuyên gia, chúng tôi khuyến cáo một chiến lược cân nhắc ICD dự phòng nguyên phát cho những trẻ em BCTPĐ có ≥ 1 yếu tố nguy cơ đột tử do tim chính với việc hiểu rằng mức độ gia tăng nguy cơ khi chỉ có một yếu tố nguy cơ đơn độc là không rõ và nguy cơ có thể cao hơn khi có nhiều yếu tố nguy cơ cùng xuất hiện trên một bệnh nhân Hình 3 và Bảng 4).
Dầy thất trái nặng: Có mối tương quan giữa sự gia tăng độ dầy thành sau thất trái và độ dầy vách liên thất (Z-score) với nguy cơ đột tử do tim ở trẻ em(92,104). Mặc dù độ dầy thành thất có tương quan với gia tăng nguy cơ đột tử do tim, mối tương quan này là một đường cong và nguy cơ có vẻ tối đa khi Z-score xấp xỉ 20. Các nghiên cứu xác định một giá trị Z-score thấp hơn ở mức trên 6 như là một yếu tố nguy cơ cao chủ yếu dựa trên mối tương quan với tiêu chí kết hợp tử vong do tim hoặc cấy ghép tim hơn là tiêu chí đột tử do tim đơn độc(105). Do đó chúng tôi thống nhất rằng một giá trị Z-score bằng 6 là thấp một cách không thích hợp và có thể phân loại quá mức trẻ em vào nhóm có nguy cơ cao đột tử do tim.
Ngất chưa rõ nguyên nhân: Đánh giá dựa theo tiền sử bệnh nhân rằng cơn ngất có vẻ không do nguyên nhân thần kinh-tim (ngất do phản xạ thần kinh phế vị), cơn ngất chưa rõ nguyên nhân có mối tương quan mạnh mẽ với nguy cơ đột tử do tim ở những bệnh nhân trẻ em BCTPĐ.
Tiền sử gia đình đột tử do tim sớm do BCTPĐ: Ở những bệnh nhân trẻ em, dữ liệu về tiền sử gia đình có người đột tử do tim mâu thuẫn với nhau và nhiều nghiên cứu không tìm ra mối tương quan với đột tử do tim ở trẻ em. Tuy nhiên, dữ liệu từ các nghiên cứu này có thể bị nhiễu bởi không xác định được đầy đủ hồ sơ di truyền của bệnh nhân (biến thể mới xuất hiện so với biến thể gia đình), mối quan hệ với bệnh nhân và tuổi xuất hiện đột tử do tim ở các thành viên trong gia đình. Đột tử do tim ở các thành viên gia đình có thể phù hợp hơn nếu đột tử xảy ra ở tuổi rất trẻ (như thời thơ ấu hoặc những năm tuổi thanh thiếu niên) hoặc nếu đột tử do tim xảy ra ở nhiều thành viên trong gia đình.
Nhịp nhanh thất không kéo dài: Nhịp nhanh thất không kéo dài được quan sát qua máy theo dõi điện tâm đồ lưu động 24 đến 48 giờ có liên quan tới gia tăng nguy cơ đột tử do tim và mối tương quan mạnh mẽ hơn như là một yếu tố nguy cơ độc lập ở những bệnh nhân BCTPĐ trẻ hơn. Vì tần số nhịp xoang bình thường ở trẻ em có thể vượt qua tần số nhanh thất theo hướng dẫn dành cho người lớn, nhịp nhanh thất ở trẻ em được định nghĩa khi tần số thất vượt quá 20% so với tần số nhịp xoang hiệu chỉnh theo tuổi.
Những điểm khác cần xem xét: Các nghiên cứu đa trung tâm gần đây báo cáo rằng chỉ số Z-score của kích thước nhĩ trái có mối tương quan dương(96,106), trong khi chênh áp đường ra thất trái khi nghỉ không có tương quan với nguy cơ đột tử do tim ở trẻ em(83,92). Điểm số ước tính nguy cơ kết hợp các yếu tố nguy cơ cùng với đường kính nhĩ trái hiệu chỉnh Z-score đã được phát triển ở những bệnh nhân BCTPĐ trẻ em nhưng vẫn chưa được sử dụng tiến cứu trong việc đưa ra quyết định cấy ICD trên lâm sàng. Mặc dù rối loạn chức năng tâm thu thất trái và phình mỏm không thường gặp ở trẻ em, nên xem xét những đặc điểm này dựa trên bằng chứng của bệnh nhân trưởng thành là có khả năng gia tăng nguy cơ đột tử do tim ở trẻ em nhưng cần phải đặt trong bối cảnh toàn bộ hồ sơ nguy cơ của từng bệnh nhân. Cuối cùng, sự phức tạp và tác động tâm lý tiềm ẩn của việc đưa ra quyết định cấy ICD ở nhóm tuổi này cần được nhấn mạnh, do thời gian dài tiếp xúc với liệu pháp ICD ở những bệnh nhân trẻ và tỷ lệ biến chứng cao hơn tương đối ở nhóm bệnh nhân này.
Ở những bệnh nhân BCTPĐ ≥ 16 tuổi với ≥ 1 yếu tố nguy cơ đột tử do tim chính, ước tính nguy cơ đột tử do tim 5 năm có thể giúp bệnh nhân hiểu được nguy cơ đột tử do tim của họ để hỗ trợ việc đưa ra quyết định cấy ICD.3,19 Trẻ em tuổi từ 16 đến 18 chiếm 2% trong nhóm đoàn hệ sử dụng để tính toán nguy cơ ở bệnh nhân trưởng thành. Tỷ lệ đại diện thấp của nhóm tuổi này nên được cân nhắc trong việc ước tính nguy cơ với những bệnh nhân trong nhóm tuổi này.
Vùng LGE lan rộng ở nhiều vùng của thất trái có tương quan với gia tăng nguy cơ rối loạn nhịp thất đe dọa tính mạng trong tương lai ở bệnh nhân trưởng thành, độc lập với vị trí cũng như hình dạng vùng LGE trong thành thất trái (82–84) Một số nghiên cứu đã đưa ra giá trị vùng LGE rộng ≥ 15% khối cơ thất trái thể hiện sự gia tăng đáng kể nguy cơ đột tử do tim(83,84); tuy nhiên, có nhiều phương pháp được sử dụng để định lượng LGE, có thể dẫn tới các kết quả khác nhau và chưa có được sự đồng thuận về phương pháp nào là tối ưu. Mối tương quan cắt ngang mạnh mẽ giữa LGE và nhịp nhanh thất không kéo dài ở những bệnh nhân BCTPĐ cung cấp thêm bằng chứng hỗ trợ cho LGE thể hiện cấu trúc bệnh lý nền tảng của rối loạn nhịp nhanh thất trong BCTPĐ. Ngoài ra, sự gia tăng số cơn nhịp nhanh thất không kéo dài quan sát trên máy theo dõi điện tâm đồ lưu động 24 đến 48 giờ cũng có tương quan với sự gia tăng nguy cơ đột tử do tim, đặc biệt là khi cơn nhịp nhanh thất không kéo dài thường xuyên, kéo dài và tần số nhanh(78) được xem là một yếu tố nguy cơ độc lập có nặng ký nhất ở bệnh nhân BCTPĐ trưởng thành. Khi không có sự tác động của các dấu ấn nguy cơ chính khác, tác động của những cơn nhịp nhanh thất không kéo dài ngắn, đơn độc đối với nguy cơ đột tử do tim ít chắc chắn hơn(78,96). Lợi ích của theo dõi điện tâm đồ kéo dài hơn với các thiết bị theo dõi lưu động dài hạn hơn cho mục đích phân tầng nguy cơ trong BCTPĐ vẫn chưa rõ ràng.
Tương quan giữa nguy cơ đột tử do tim và LGE ở những trẻ em BCTPĐ vẫn chưa được xác định rõ. Mặc dù gần một nửa các trẻ em lớn và trẻ vị thành niên có LGE, mức độ lan rộng của vùng LGE xác định nguy cơ cao ở trẻ em vẫn chưa được xác định(107,108), Tuy nhiên, do LGE thể hiện cấu trúc bệnh lý nền tảng của nhịp nhanh thất làm tăng nguy cơ đột tử do tim ở bệnh nhân BCTPĐ trưởng thành(82–84),có vẻ thích hợp khi cân nhắc sự mở rộng vùng LGE có khả năng làm tăng nguy cơ đột tử do tim ở trẻ em. Rối loạn chức năng tâm thu ít gặp ở trẻ em, nhưng có vẻ cũng gia tăng nguy cơ biến cố bất lợi, bao gồm cả đột tử do tim. Có thể cần thuốc an thần hoặc gây mê toàn thân để thu được hình ảnh cộng hưởng từ tim ở những bệnh nhân trẻ.
Do các biến chứng dài hạn liên quan đến việc đặt ICD, không nên áp dụng liệu pháp dùng thiết bị ở những bệnh nhân BCTPĐ không có bằng chứng gia tăng nguy cơ đột tử do tim dựa trên thuật toán yếu tố nguy cơ được đề xuất(109,110) (Hình 3).
Sẽ là không thích hợp để khuyến cáo liệu pháp ICD ở những bệnh nhân BCTPĐ có hồ sơ lâm sàng nguy cơ thấp đột tử do tim, chỉ vì mục đích duy nhất là cho phép bệnh nhân trở lại các môn thể thao cạnh tranh(111).
5.3 Cân nhắc lựa chọn thiết bị
| Loại | MCC | Khuyến cáo về lựa chọn loại ICD |
| 1 | B-NR | 1. Ở những bệnh nhân BCTPĐ được cấy ICD, cả ICD một buồng đường tĩnh mạch và ICD dưới da được khuyến cáo sau khi thảo luận về việc đưa ra quyết định có cân nhắc tới sở thích, lối sống của bệnh nhân và nhu cầu cần tạo nhịp cho nhịp chậm hoặc cắt cơn nhịp nhanh thất. |
| 1 | B-NR | 2. Ở những bệnh nhân BCTPĐ được cấy ICD, điện cực ICD một cuộn dây điện từ (coil) được khuyến cáo ưu tiên hơn so với dùng điện cực hai cuộn dây điện từ (coil)(112). |
| 2a | B-NR | 3. Ở những bệnh nhân BCTPĐ được cấy ICD, ICD hai buồng được cân nhắc với những bệnh nhân cần tạo nhịp nhĩ hoặc tạo nhịp nhĩ và thất tuần tự vì rối loạn nhịp chậm hoặc rối loạn dẫn truyền, hoặc như một nỗ lực để làm giảm các triệu chứng của BCTPĐ có tắc nghẽn (thưởng ở những bệnh nhân trên 65 tuổi). |
| 2a | C-LD | 4. Ở nhóm bệnh nhân BCTPĐ không tắc nghẽn trưởng thành được cấy ICD có triệu chứng suy tim NYHA II-IV, blốc nhánh trái và phân suất tống máu thất trái dưới 50%, điều trị tái đồng bộ tim (CRT) để cải thiện triệu chứng nên được cân nhắc (113–115). |
| 2b | C-LD | 5. Ở những bệnh nhân BCTPĐ đã đưa ra quyết định cấy ICD và có nhịp nhanh nhĩ hoặc rung nhĩ kích phát, máy ICD hai buồng có thể được cân nhắc, nhưng quyết định này cần cân bằng với tỷ lệ biến chứng cao hơn ở các thiết bị hai buồng. |
Nội dung khuyến cáo
Quyết định lựa chọn loại ICD để cấy là rất quan trọng và tinh tế khi cân nhắc tới các lợi ích và nguy cơ của từng loại thiết bị. Việc cân nhắc bao gồm ICD đường tĩnh mạch hay cấy dưới da, tạo nhịp một buồng hay hai buồng hoặc CRT và số lượng cuộn dây điện từ khử rung khi tiếp cận theo đường tĩnh mạch. Những bệnh nhân BCTPĐ được cấy ICD thường trẻ hơn so với những bệnh nhân được cấy ICD ở nhóm bệnh tim thiếu máu cục bộ hoặc các bệnh cơ tim không do thiếu máu cục bộ, vì vậy nên tỷ lệ biến chứng lâu dài có vẻ cao hơn ở nhóm BCTPĐ.
Cân nhắc ở trẻ em
Lựa chọn cấy ICD ở trẻ em là một thách thức(116–118). Mặc dù việc lựa chọn bệnh nhân nên được cấy ICD được thảo luận tại phần trước, chiến lược cấy sẽ thay đổi tùy theo kích thước cơ thể của bệnh nhân. Điện cực ngoại mạc thường cần thiết ở những trẻ nhỏ hơn, thường là dưới 30 kg và những trẻ cần đặt điện cực thất trái / CRT. Biến chứng của ICD có thể cao hơn ở trẻ em và trẻ vị thành niên bởi nhịp tim cơ bản cao hơn có thể dẫn tới những lần sốc không thích hợp, sự tăng trưởng của trẻ làm gia tăng nguy cơ tổn thương điện cực và cần nhiều lần thay máy / tháo máy trong suốt cuộc đời(116). Ở những bệnh nhân trẻ hơn, điện cực đường tĩnh mạch cho thấy tỷ lệ hỏng cao hơn so với bệnh nhân cao tuổi. Một nhóm nhỏ bệnh nhân cấy ICD dưới da cũng có thể có nguy cơ biến chứng cao hơn, bao gồm cả sự ăn mòn của thiết bị(117–119).
- Quyết định về việc cấy ICD bao gồm nhiều vấn đề cần cân nhắc, bao gồm ICD đường tĩnh mạch hay dưới da, một buồng hay hai buồng hay CRT và số lượng cuộn dây điện từ khử rung. Lợi ích của thiết bị đường tĩnh mạch là có thể tạo nhịp khi nhịp chậm và khả năng tạo nhịp từ mỏm thất phải để giảm triệu chứng, kích thước máy nhỏ, pin tuổi thọ dài và kinh nghiệm sử dụng lâu hơn. Điều bất lợi là điện cực có thể bị hư dần theo thời gian, cần đặt thêm các điện cực mới và loại bỏ điện cực cũ, đi kèm với việc tăng nguy cơ. Ngoài ra, nhiễm trùng máy và điện cực có thể đưa đến viêm nội tâm mạc nhiễn trùng. Điểm mạnh của ICD dưới da là không có điện cực tĩnh mạch, nguy cơ hỏng điệc cực thấp hơn và dễ gỡ bỏ. Điểm bất lợi bao gồm kích thước thiết bị lớn, thời gian dùng pin ngắn hơn, nguy cơ sốc không thích hợp tăng lên bởi nhận cảm quá mức sóng T và sự run cơ và thời gian sử dụng máy thấp hơn. Những bệnh nhân BCTPĐ được cấy ICD dưới da nên được đánh giá nguy cơ nhận cảm quá mức sau khi gắng sức và cả thời gian sử dụng ngắn hơn. Những bệnh nhân BCTPĐ được cấy ICD dưới da nên được tầm soát hiện tượng nhận cảm quá mức sau khi gắng sức và có thể phải làm trắc nghiệm gắng sức sau khi cấy ICD.
- Việc thảo luận để cùng đưa ra quyết định cần tính đến lối sống, sở thích của bệnh nhân và dự đoán nguy cơ cần tạo nhịp cho nhịp chậm hoặc chấm dứt cơn nhanh thất. Chúng ta cần xem xét tuổi của bệnh nhân, nguy cơ cần tạo nhịp do nhịp chậm và cân nhắc về những lần sốc không thích hợp và độ bền của điện cực.
- Hệ thống một buồng có ít biến chứng hơn cả trong theo dõi ngắn hạn và dài hạn khi so sánh với hệ thống hai buồng đường tĩnh mạch. Do đó, các thiết bị một buồng thường được ưu tiên hơn các hệ thống hai buồng.
- Điện cực ICD một cuộn dây điện từ có ít biến chứng hơn khi tháo ra nhưng kèm theo nguy cơ tăng ngưỡng khử rung. Tuy nhiên, hầu hết bệnh nhân cả ở nhóm mắc BCTPĐ hoặc không, đều có biên độ an toàn đầy đủ với điện cực một cuộn dây điện từ. Các điện cực một cuộn dây điện từ được sử dụng hầu hết khi cấy máy bên vai trái, và các dữ liệu từ nhóm dân số không mắc BCTPĐ gợi ý rằng các điện cực hai cuộn dây điện từ là cần thiết khi cấy bên vai phải. Do đó, khuyến cáo đối với điện cực một cuộn dây điện từ chỉ áp dụng khi cấy máy bên vai trái. Cuối cùng, cần chú ý mạnh mẽ tới ngưỡng khử rung ở những bệnh nhân có điện cực một cuộn dây điện từ, cấy máy bên vai phải hoặc cơ tim phì đại nặng.
- Ở những bệnh nhân BCTPĐ cần tạo nhịp nhĩ, hệ thống hai buồng là cần thiết. Đã có bốn thử nghiệm lâm sàng ngẫu nhiên có đối chứngcho kết quả nhất quán về lợi ích của tạo nhịp thất phải ơ những bệnh nhân BCTPĐ có chênh áp đường ra thất trái ≥ 30 Trong giai đoạn cấp, tạo nhịp thất phải giảm chênh áp đường ra thất trái, nhưng lợi ích lâm sàng về dài hạn vẫn chưa được chứng minh.
- Mặc dù hầu hết các bằng chứng ủng hộ lợi ích của CRT từ các nghiên cứu có rất ít hoặc không có bệnh nhân BCTPĐ, sẽ là hợp lý khi chúng ta sử dụng phương pháp này với những bệnh nhân BCTPĐ khi đã đáp ứng đủ tiêu chẩn CRT theo các hướng dẫn hiện tại(120) trong suy tim, bao gồm những bệnh nhân có mức NYHA II – IV, phân suất tống máu ≤ 35% và QRS dãn rộng. Những bệnh nhân có blốc nhánh trái và QRS ≥ 150 ms được khuyến cáo loại 1, trong khi những bệnh nhân có blốc nhánh trái và QRS từ 120 – 149 ms và những bệnh nhân không có blốc nhánh trái và QRS dãn rộng ≥ 150 ms được khuyến cáo loại 2a và những bệnh nhân không có blốc nhánh trái và QRS dãn rộng 120 – 149 ms được khuyến cáo loại 2b. Ngoài những bệnh nhân kể trên, chúng ta đã có một số nghiên cứu nhỏ về CRT-D ở những bệnh nhân BCTPĐ và phân suất tống máu trên 35% (113–115,121,122). Khoảng một nửa số bệnh nhân có đáp ứng lâm sàng với CRT qua việc cải thiện mức độ NYHA, hoặc có bằng chứng đảo ngược tái cấu trúc thất. Lợi ích có vẻ lớn hơn ở bệnh nhân có LBB và QRS rất rộng. Những người đáp ứng cho thấy có sự cải thiện khiêm tốn phân suất tống máu thất trái. Một nghiên cứu khác cho thấy CRT giúp kéo dài đáng kể thời gian cần đặt dụng cụ hỗ trợ thất trái, ghép tim hoặc tử vong(115), trong khi hai nghiên cứu khác không ghi nhận sự cải thiện sống còn(113–115,121,122). Tạo nhịp thất phải có cùng hình ảnh blốc nhánh trái nên khuyến cáo này có thể mở rộng đối với những bệnh nhân phân suất tống máu từ 35 – 50% và ước tính tạo nhịp chiếm trên 40% thời gian, tương tự như hướng dẫn về tạo nhịp AHA / ACC / HRS (2021).
- Một điện cực nhĩ có thể giúp phân biệt tốt hơn giữa nhịp nhanh thất và nhịp nhanh nhĩ, mặc dù dữ liệu còn khiêm tốn về việc giảm tỷ lệ sốc không thích hợp ở những bệnh nhân đặt máy phá rung cấy được hai buồng (ICD) và có dữ liệu về tỷ lệ biến chứng của các thiết bị hai buồng là cao hơn(123–128). Tuy nhiên, ở những bệnh nhân trẻ em có rối loạn nhịp nhanh nhĩ (tần số có thể đạt tới 180), tần số cơn nhịp nhanh có thể gần với cơn nhanh thất, máy ICD hai buồng có khả năng phân biệt nhịp nhanh trên thất và nhịp nhanh thất. Lợi thế tiềm năng này phải được cân nhắc với nguy cơ biến chứng cao hơn khi cấy máy hai buồng.
6. Điều trị bệnh nhân bệnh cơ tim phì đại
6.1 Điều trị bệnh nhân BCTPĐ tắc nghẽn
6.1.1 Điều trị bệnh nhân BCTPĐ tắc nghẽn có triệu chứng
Điều trị bằng thuốc ở bệnh nhân BCTPĐ tắc nghẽn có triệu chứng
| Loại | MCC | Khuyến cáo điều trị bằng thuốc ở bệnh nhân BCTPĐ tắc nghẽn |
| 1 | B-NR | 1. Bệnh nhân BCTPĐ tắc nghẽn có triệu chứng* do nghẽn đường ra thất trái, chẹn beta không dãn mạch được khuyến cáo, tăng dần tới liều có hiệu quả hoặc liều tối đa có thể dung nạp(129–131). |
| 1 | Verapamil
B-NR |
2. Bệnh nhân BCTPĐ tắc nghẽn có triệu chứng* do nghẽn đường ra thất trái, khi chẹn beta không hiệu quả hoặc không dung nạp, khuyến cáo thay thế bằng chẹn calci nhóm non-dihydropyridine (verapamil, diltiazem) (132–134). |
| Diltiazem
C-LD |
||
| 1 | B-NR | 3. Bệnh nhân BCTPĐ tắc nghẽn có triệu chứng* do nghẽn đường ra thất trái mặc dù đã điều trị với chẹn beta hoặc chẹn calci non-dihydropyridine, khuyến cáo bổ sung disopyramide kết hợp một trong những nhóm thuốc khác; hoặc chỉ định điều trị giảm độ dầy vách liên thất tại những trung tâm chuyên sâu(11,135–139). |
| 1 | C-LD | 4. Bệnh nhân BCTPĐ tắc nghẽn và hạ huyết áp cấp tính không đáp ứng với dịch truyền, khuyến cáo phenylephrine tĩnh mạch (hoặc những thuốc co mạch khác không có tác dụng tăng co cơ tim), dùng đơn độc hoặc kết hợp chẹn beta(140). |
| 2b | C-EO | 5. Bệnh nhân BCTPĐ tắc nghẽn và khó thở dai dẳng có bằng chứng lâm sàng của quả tải thể tích tuần hoàn và áp lực đổ đầy tim trái cao mặc dù đã điều trị tối ưu theo hướng dẫn của khuyến cáo, có thể cân nhắc sử dụng lợi tiểu uống liều thấp một cách thận trọng. |
| 2b | C-EO | 6. Bệnh nhân BCTPĐ tắc nghẽn, việc ngưng sử dụng những thuốc dãn mạch (như ức chế men chuyển, chẹn thụ thể angiotensin, chẹn calci nhóm dihydropyridine) hoặc digoxin có thể phù hợp, do những thuốc này có thể làm nặng thêm triệu chứng gây ra bởi tắc nghẽn buồng tống động học. |
| 2b | B-R | 7. Mavacamten có thể được điều trị làm giảm triệu chứng và trì hoãn việc can thiệp giảm độ dầy vách thất. |
| 3: có hại | C-LD | 8. Bệnh nhân BCTPĐ tắc nghẽn và khó thở nặng lúc nghỉ, hạ huyết áp, chênh áp khi nghỉ rất cao (trên 100 mmHg) cũng như trẻ em dưới 6 tuần tuổi, verapamil có thể có hại. |
| * Triệu chứng: khó thở hoặc đau ngực có liên quan gắng sức; và đôi khi triệu chứng khác liên quan gắng sức (như ngất, gần ngất) do nghẽn đường ra thất trái và làm ảnh hưởng đến hoạt động thông thường hàng ngày của bệnh nhân hoặc chất lượng cuộc sống. | ||
Nội dung khuyến cáo
Vai trò chính của điều trị bằng thuốc là nhằm cải thiện tắc nghẽn thất trái động học để giảm triệu chứng, chưa có dữ liệu đủ thuyết phục điều trị bằng thuốc cải thiện tiên lượng BCTPĐ. Do mức độ tắc nghẽn đường ra thất trái thay đổi đáng kể trong cuộc sống hàng ngày của bệnh nhân, nên sự thành công của một thuốc điều trị cụ thể được xác định bởi đáp ứng triệu chứng của bệnh nhân chứ không phải độ chênh áp đo được. Nhìn chung, chẹn beta không dãn mạch được xem là điều trị hàng đầu. Chẹn calci, verapamil, hoặc diltiazem, là những sự thay thế phù hợp nếu chẹn beta không thể chỉ định. Đối với những bệnh nhân không đáp ứng điều trị với một loại thuốc kể trên, những điều trị chuyên sâu hơn với disopyramide hoặc làm giảm độ dầy vách liên thất thường là bước tiếp theo. Một trong những bước quan trọng khác trong điều trị triệu chứng BCTPĐ tắc nghẽn là tránh những thuốc có thể làm nặng thêm tắc nghẽn đường ra thất trái, như dãn mạch đơn thuần (ví dụ: chẹn calci nhóm dihydropyridine, ức chế men chuyển, chẹn thụ thể angiotensin) và liều cao lợi tiểu. Liều thấp lợi tiểu, khi được thêm vào những điều trị hàng đầu khác có thể hữu ích đối với những bệnh nhân có khó thở hay triệu chứng sung huyết phổi. Nguyên tắc chính của điều trị bằng thuốc được liệt kê dưới đây bao gồm cho những bệnh nhân tắc nghẽn ở giữa buồng thất.
- Chẹn beta là thuốc điều trị cho tắc nghẽn buồng tống động học được nghiên cứu đầu tiên và nhìn chung là điều trị đầu tay đối với hầu hết bệnh nhân BCTPĐ tắc nghẽn. Thuốc nên được tăng liều dần tới khi đạt được cải thiện triệu chứng, không nên gọi là thất bại của chẹn thụ thể beta cho tới khi có bằng chứng tác dụng của chẹn beta được chứng minh (ví dụ như ức chế tần số tim lúc nghỉ)(129–131).
- Diltiazem và verapamil đều được chứng minh là làm giảm được triệu chứng ở những bệnh nhân BCTPĐ tắc nghẽn. Cả hai thuốc này đều có một số giới hạn như: tính dãn mạch, tác dụng tăng co cơ tim âm và tăng tần số tim âm tính. Sử dụng chẹn calci kết hợp với chẹn beta như là một hướng dẫn cho điều trị BCTPĐ, nhưng chưa có bằng chứng ủng hộ phối hợp này (132–134); Tuy nhiên, điều trị này có thể có vai trò ở bệnh nhân tăng huyết áp kèm theo.
- Những bệnh nhân BCTPĐ không đáp ứng với chẹn beta hoặc chẹn calci nhóm non-dihydropyridine cần những điều trị chuyên sâu hơn, bao gồm disopyramide và điều trị làm giảm độ dầy vách liên thất được thực hiện bởi thủ thuật viên có kinh nghiệm tại những trung tâm chuyên sâu. Việc lựa chọn giữa những phương pháp này nên được tiếp cận thông qua thảo luận với bệnh nhân bao gồm tỷ lệ thành công, lợi ích, nguy cơ của mỗi phương pháp. Disopyramide đã cho thấy được lợi ích về mặt triệu chứng ở những bệnh nhân BCTPĐ tắc nghẽn thất bại với điều trị chẹn beta, verapamil hoặc diltiazem(135–137). Thuốc này là một chọn lựa quan trọng, đặc biệt ở những bệnh nhân không có chỉ định điều trị giảm độ dầy vách liên thất. Do disopyramide có thể làm tăng dẫn truyền thông qua nút nhĩ thất, có thể dẫn tới dẫn truyền rất nhanh khi có cơn rung nhĩ, thuốc này nên được dùng kết hợp với những thuốc khác có khả năng ức chế nút nhĩ thất (chẹn beta, verapamil hoặc diltiazem). Tác dụng phụ đối giao cảm có thể xảy ra ở disopyramide có thể bị nhầm lẫn với pyridostigmine. Ở những bệnh nhân BCTPĐ tắc nghẽn vẫn còn triệu chứng nặng mặc dù đã điều trị nội tối ưu, điều trị giảm độ dầy vách liên thất, được thực hiện bởi những thủ thuật viên có kinh nghiệm tại trung tâm chuyên sâu, rất hiệu quả trong làm giảm tắc nghẽn đường ra thất trái(138). Sống còn ở những bệnh nhân có tắc nghẽn đường ra thất trái giảm so với bệnh nhân không tắc nghẽn, nên việc giảm tình trạng tắc nghẽn có thể làm giảm nguy cơ tử vong(139,141).
- Hạ huyết áp cấp tính ở những bệnh nhân BCTPĐ tắc nghẽn là cấp cứu nội khoa. Tối đa hoá tiền tải và hậu tải, tránh làm tăng co bóp cơ tim hoặc tần số tim, là điểm quan trọng trong điều trị hạ huyết áp cấp tính. Truyền tĩnh mạch những thuốc co mạch, ví dụ như phenylephrine, có thể làm đảo ngược tình trạng nguy hiểm này. Chẹn beta cũng có thể hữu ích khi kết hợp với thuốc co mạch do nó làm giảm co bóp cơ tim và cải thiện tiền tải bằng cách kéo dài thời gian đổ đầy tâm trương.
- Khi có dấu hiệu hoặc triệu chứng sung huyết, việc sử dụng một cách thận trọng lợi tiểu liều thấp có thể cải thiện triệu chứng. Tuy nhiên, lợi niệu quá tích cực có thể là một vấn đề, do làm giảm tiền tải và có thể làm tăng thêm nghẽn đường ra thất trái.
- Cần phải thận trọng khi sử dụng thuốc ở bệnh nhân BCTPĐ có điều trị những bệnh lý đồng mắc. Một vài thuốc điều trị có thể gây ra hoặc làm nặng thêm triệu chứng tắc nghẽn đường ra thất trái. Ví dụ sử dụng lợi tiểu và dãn mạch để điều trị tăng huyết áp hoặc bảo vệ chức năng thận. Những thuốc này có thể dùng ở những bệnh nhân không triệu chứng. Tuy nhiên nếu triệu chứng xuất hiện, hoặc khởi phát sau khi khởi trị thuốc, có thể cần phải tăng liều những thuốc dùng để điều trị cho BCTPĐ tắc nghẽn hoặc cân nhắc những trị liệu thay thế cho những tình trạng bệnh lý đồng mắc. Do đó, những thuốc có tác dụng tăng co cơ tim dương, dãn mạch đơn thuần và lợi tiểu liều cao là chống chỉ định tương đối ở những bệnh nhân BCTPĐ tắc nghẽn có triệu chứng.
- Mặc dù verapamil và diltiazem là những thuốc điều trị để làm giảm triệu chứng liên quan đến tắc nghẽn đường ra thất trái, một vài bệnh nhân đã được ghi nhận hoạt tính dãn mạch nổi trội của thuốc này. Tác dụng giảm hậu tải này có thể đặc biệt nguy hiểm ở những bệnh nhân với chênh áp lúc nghỉ rất cao (trên 80 tới 100 mmHg) và có dấu hiệu của suy tim sung huyết. Đã có một vài báo cáo của nhịp chậm đe doạ tính mạng và tụt huyết áp ở trẻ mới sinh dưới 6 tuần tuổi điều trị nhịp nhanh trên thất với verapamil đường tĩnh mạch(142). Tuy nhiên, verapamil cho thấy hiệu quả và dung nạp tốt khi sử dụng ở trẻ sơ sinh và trẻ nhỏ lớn hơn với BCTPĐ ở trong điều kiện kiểm soát(143).
Các kết quả của các nghiên cứu lâm sàng gần đây về nhóm thuốc ức chế myosin tim (Mavacamten) trong điều trị bệnh nhân BCTPĐ tắc nghẽn: MAVA-LTE, EXPLORER-LTE(144), EXPLORER-HCM(145), VALOR-HCM(146). Cho thấy cải thiện triệu chứng ngắn, trung hạn cũng như có thể trì hoãn can thiệp giảm độ dầy vách liên thất bệnh nhân BCTPĐ tắc nghẽn. Thuốc được phê chuẩn bởi FDA cho chỉ định điều trị giảm triệu chứng và tăng khả năng gắng sức ở người lớn BCTPĐ tắc nghẽn, dạng viên Camzyos (mavacamten, 2,5 mg, 5 mg, 10 mg, 15 mg) vào 28 tháng Tư năm 2022.
Xem tiếp kỳ sau…
TÀI LIỆU THAM KHẢO
- Geske JB, Ommen SR, Gersh BJ. Hypertrophic Cardiomyopathy: Clinical Update. JACC: Heart Failure. 2018;6(5):364-375. https://doi.org/10.1016/J.JCHF.2018.02.010
- Marian AJ, Braunwald E. Hypertrophic Cardiomyopathy. Circulation Research. 2017;121(7):749-770. https://doi.org/10.1161/CIRCRESAHA.117.311059
- Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic Cardiomyopathy: Present and Future, With Translation Into Contemporary Cardiovascular Medicine. J Am Coll Cardiol. 2014;64(1):83-99. https://doi.org/10.1016/J.JACC.2014.05.003
- Maron BJ. Clinical Course and Management of Hypertrophic Cardiomyopathy. New England Journal of Medicine. 2018;379(7):655-668. https://doi.org/10.1056/NEJMRA1710575
- Semsarian C, Ingles J, Maron MS, Maron BJ. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. https://doi.org/10.1016/J.JACC.2015.01.019
- Maron M, Hellawell J, cardiology JL… journal of, 2016 undefined. Occurrence of clinically diagnosed hypertrophic cardiomyopathy in the United States. Elsevier. Accessed May 4, 2022. https://www.sciencedirect.com/science/article/pii/S0002914916303046
- Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and Mechanistic Insights Into the Genetics of Cardiomyopathy. J Am Coll Cardiol. 2016;68(25):2871-2886. https://doi.org/10.1016/J.JACC.2016.08.079
- Ingles J, Burns C, Bagnall RD, et al. Nonfamilial Hypertrophic Cardiomyopathy. Circulation: Cardiovascular Genetics. 2017;10(2). https://doi.org/10.1161/CIRCGENETICS.116.001620
- Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138(14):1387-1398. https://doi.org/10.1161/CIRCULATIONAHA.117.033200
- Maron B, Rowin E, Casey S, cardiology MMJ, 2016 undefined. How hypertrophic cardiomyopathy became a contemporary treatable genetic disease with low mortality: shaped by 50 years of clinical research and practice. jamanetwork.com. Accessed May 4, 2022. https://jamanetwork.com/journals/jamacardiology/article-abstract/2498963
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114(21):2232-2239. https://doi.org/10.1161/CIRCULATIONAHA.106.644682
- Sorajja P, Nishimura RA, Gersh BJ, et al. Outcome of Mildly Symptomatic or Asymptomatic Obstructive Hypertrophic Cardiomyopathy. A Long-Term Follow-Up Study. J Am Coll Cardiol. 2009;54(3):234-241. https://doi.org/10.1016/J.JACC.2009.01.079
- Pellikka PA, Oh JK, Bailey KR, Nichols BA, Monahan KH, Tajik AJ. Dynamic intraventricular obstruction during dobutamine stress echocardiography: A new observation. Circulation. 1992;86(5):1429-1432. https://doi.org/10.1161/01.CIR.86.5.1429
- Villemain O, Correia M, Mousseaux E, et al. Myocardial Stiffness Evaluation Using Noninvasive Shear Wave Imaging in Healthy and Hypertrophic Cardiomyopathic Adults. JACC: Cardiovascular Imaging. 2019;12(7):1135-1145. https://doi.org/10.1016/J.JCMG.2018.02.002
- Paulus WJ, Lorell BH, Craig WE, Wynne J, Murgo JP, Grossman W. Comparison of the effects of nitroprusside and nifedipine on diastolic properties in patients with hypertrophic cardiomyopathy: Altered left ventricular loading or improved muscle inactivation? J Am Coll Cardiol. 1983;2(5):879-886. https://doi.org/10.1016/S0735-1097(83)80235-6
- Chan RH, Maron BJ, Olivotto I, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484-495. https://doi.org/10.1161/CIRCULATIONAHA.113.007094
- Sherrid M v., Balaram S, Kim B, Axel L, Swistel DG. The Mitral Valve in Obstructive Hypertrophic Cardiomyopathy: A Test in Context. J Am Coll Cardiol. 2016;67(15):1846-1858. https://doi.org/10.1016/J.JACC.2016.01.071
- Maron MS, Olivotto I, Harrigan C, et al. Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011;124(1):40-47. https://doi.org/10.1161/CIRCULATIONAHA.110.985812
- Hodges K, Rivas CG, Aguilera J, et al. Surgical management of left ventricular outflow tract obstruction in a specialized hypertrophic obstructive cardiomyopathy center. The Journal of Thoracic and Cardiovascular Surgery. 2019;157(6):2289-2299. https://doi.org/10.1016/J.JTCVS.2018.11.148
- Hong JH, Schaff H v., Nishimura RA, et al. Mitral Regurgitation in Patients With Hypertrophic Obstructive Cardiomyopathy: Implications for Concomitant Valve Procedures. J Am Coll Cardiol. 2016;68(14):1497-1504. https://doi.org/10.1016/J.JACC.2016.07.735
- Rowin EJ, Maron BJ, Haas TS, et al. Hypertrophic Cardiomyopathy With Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J Am Coll Cardiol. 2017;69(7):761-773. https://doi.org/10.1016/J.JACC.2016.11.063
- Patel V, Critoph CH, Finlay MC, Mist B, Lambiase PD, Elliott PM. Heart Rate Recovery in Patients With Hypertrophic Cardiomyopathy. The American Journal of Cardiology. 2014;113(6):1011-1017. https://doi.org/10.1016/J.AMJCARD.2013.11.062
- Olivotto I, Maron BJ, Montereggi A, Mazzuoli F, Dolara A, Cecchi F. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999;33(7):2044-2051. https://doi.org/10.1016/S0735-1097(99)00094-7
- Frenneaux MP, Counihan PJ, Caforio ALP, Chikamori T, McKenna WJ. Abnormal blood pressure response during exercise in hypertrophic cardiomyopathy. Circulation. 1990;82(6):1995-2002. https://doi.org/10.1161/01.CIR.82.6.1995
- Sadoul N, Prasad K, Elliott PM, Bannerjee S, Frenneaux MP, McKenna WJ. Prospective Prognostic Assessment of Blood Pressure Response During Exercise in Patients With Hypertrophic Cardiomyopathy. Circulation. 1997;96(9):2987-2991. https://doi.org/10.1161/01.CIR.96.9.2987
- Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):e558-e631. https://doi.org/10.1161/CIR.0000000000000937
- Ahmad F, McNally EM, Ackerman MJ, et al. Establishment of Specialized Clinical Cardiovascular Genetics Programs: Recognizing the Need and Meeting Standards: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2019;12(6):286-305. https://doi.org/10.1161/HCG.0000000000000054
- Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2010;31(22):2715-2728. https://doi.org/10.1093/EURHEARTJ/EHQ271
- Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol. 2012;60(8):705-715. https://doi.org/10.1016/J.JACC.2012.02.068
- Ingles J, Sarina T, Yeates L, et al. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15(12):972-977. https://doi.org/10.1038/GIM.2013.44
- van Velzen HG, Schinkel AFL, Baart SJ, et al. Outcomes of Contemporary Family Screening in Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2018;11(4):e001896. https://doi.org/10.1161/CIRCGEN.117.001896
- Ranthe MF, Carstensen L, Øyen N, et al. Risk of Cardiomyopathy in Younger Persons With a Family History of Death from Cardiomyopathy: A Nationwide Family Study in a Cohort of 3.9 Million Persons. Circulation. 2015;132(11):1013-1019. https://doi.org/10.1161/CIRCULATIONAHA.114.013478
- Lafreniere-Roula M, Bolkier Y, Zahavich L, et al. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur Heart J. 2019;40(45):3672-3681. https://doi.org/10.1093/EURHEARTJ/EHZ396
- Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17(11):880-888. https://doi.org/10.1038/GIM.2014.205
- Bagnall RD, Ingles J, Dinger ME, et al. Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2018;72(4):419-429. https://doi.org/10.1016/J.JACC.2018.04.078
- Ingles J, Burns C, Funke B. Pathogenicity of Hypertrophic Cardiomyopathy Variants. Circulation: Cardiovascular Genetics. 2017;10(5). https://doi.org/10.1161/CIRCGENETICS.117.001916
- Maron BJ, Roberts WC, Arad M, et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA. 2009;301(12):1253-1259. https://doi.org/10.1001/JAMA.2009.371
- Desai MY, Ommen SR, McKenna WJ, Lever HM, Elliott PM. Imaging phenotype versus genotype in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2011;4(2):156-168. https://doi.org/10.1161/CIRCIMAGING.110.957936
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. https://doi.org/10.1038/GIM.2015.30
- Ouellette AC, Mathew J, Manickaraj AK, et al. Clinical genetic testing in pediatric cardiomyopathy: Is bigger better? Clin Genet. 2018;93(1):33-40. https://doi.org/10.1111/CGE.13024
- Jensen MK, Havndrup O, Christiansen M, et al. Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing. Circulation. 2013;127(1):48-54. https://doi.org/10.1161/CIRCULATIONAHA.111.090514
- Semsarian C, Ingles J, Wilde AAM. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J. 2015;36(21):1290-1296. https://doi.org/10.1093/EURHEARTJ/EHV063
- Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. New England Journal of Medicine. 2016;374(25):2441-2452.
- Jipin Das K, Ingles J, Bagnall RD, Semsarian C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet Med. 2014;16(4):286-293. https://doi.org/10.1038/GIM.2013.138
- Manrai AK, Funke BH, Rehm HL, et al. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med. 2016;375(7):655-665. https://doi.org/10.1056/NEJMSA1507092
- Mathew J, Zahavich L, Lafreniere-Roula M, et al. Utility of genetics for risk stratification in pediatric hypertrophic cardiomyopathy. Clin Genet. 2018;93(2):310-319. https://doi.org/10.1111/CGE.13157
- Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42(10). https://doi.org/10.1136/JMG.2005.033886
- Aronson SJ, Clark EH, Varugheese M, Baxter S, Babb LJ, Rehm HL. Communicating new knowledge on previously reported genetic variants. Genet Med. 2012;14(8):713-719. https://doi.org/10.1038/GIM.2012.19
- Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249-1254. https://doi.org/10.1016/J.JACC.2015.01.019
- Ingles J, Goldstein J, Thaxton C, et al. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circulation: Genomic and Precision Medicine. 2019;12(2):57-64. https://doi.org/10.1161/CIRCGEN.119.002460
- Elliott P, Baker R, Pasquale F, et al. Prevalence of Anderson-Fabry disease in patients with hypertrophic cardiomyopathy: the European Anderson-Fabry Disease survey. Heart. 2011;97(23):1957-1960. https://doi.org/10.1136/HEARTJNL-2011-300364
- Lafreniere-Roula M, Bolkier Y, Zahavich L, et al. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur Heart J. 2019;40(45):3672-3681. https://doi.org/10.1093/EURHEARTJ/EHZ396
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. https://doi.org/10.1038/GIM.2015.30
- Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332(16):1058-1065. https://doi.org/10.1056/NEJM199504203321603
- Olivotto I, Girolami F, Ackerman MJ, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83(6):630-638. https://doi.org/10.4065/83.6.630
- Captur G, Lopes LR, Mohun TJ, et al. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2014;7(6):863-871. https://doi.org/10.1161/CIRCIMAGING.114.002411
- Ho CY, Day SM, Colan SD, et al. The Burden of Early Phenotypes and the Influence of Wall Thickness in Hypertrophic Cardiomyopathy Mutation Carriers: Findings From the HCMNet Study. JAMA Cardiol. 2017;2(4):419-428. https://doi.org/10.1001/JAMACARDIO.2016.5670
- Vigneault DM, Yang E, Jensen PJ, et al. Left Ventricular Strain Is Abnormal in Preclinical and Overt Hypertrophic Cardiomyopathy: Cardiac MR Feature Tracking. Radiology. 2019;290(3):640-648. https://doi.org/10.1148/RADIOL.2018180339
- Williams LK, Misurka J, Ho CY, et al. Multilayer Myocardial Mechanics in Genotype-Positive Left Ventricular Hypertrophy-Negative Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122(10):1754-1760. https://doi.org/10.1016/J.AMJCARD.2018.08.008
- Norrish G, Jager J, Field E, et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation. 2019;140(3):184-192. https://doi.org/10.1161/CIRCULATIONAHA.118.038846
- Christiaans I, Birnie E, Bonsel GJ, et al. Manifest disease, risk factors for sudden cardiac death, and cardiac events in a large nationwide cohort of predictively tested hypertrophic cardiomyopathy mutation carriers: determining the best cardiological screening strategy. Eur Heart J. 2011;32(9):1161-1170. https://doi.org/10.1093/EURHEARTJ/EHR092
- Maurizi N, Michels M, Rowin EJ, et al. Clinical Course and Significance of Hypertrophic Cardiomyopathy Without Left Ventricular Hypertrophy. Circulation. 2019;139(6):830-833. https://doi.org/10.1161/CIRCULATIONAHA.118.037264
- Vermeer AMC, Clur SAB, Blom NA, Wilde AAM, Christiaans I. Penetrance of Hypertrophic Cardiomyopathy in Children Who Are Mutation Positive. J Pediatr. 2017;188:91-95. https://doi.org/10.1016/J.JPEDS.2017.03.033
- Gray B, Ingles J, Semsarian C. Natural history of genotype positive-phenotype negative patients with hypertrophic cardiomyopathy. Int J Cardiol. 2011;152(2):258-259. https://doi.org/10.1016/J.IJCARD.2011.07.095
- Maron MS, Rowin EJ, Wessler BS, et al. Enhanced American College of Cardiology/American Heart Association Strategy for Prevention of Sudden Cardiac Death in High-Risk Patients With Hypertrophic Cardiomyopathy. JAMA Cardiol. 2019;4(7):644-657. https://doi.org/10.1001/JAMACARDIO.2019.1391
- O’Mahony C, Jichi F, Ommen SR, et al. International External Validation Study of the 2014 European Society of Cardiology Guidelines on Sudden Cardiac Death Prevention in Hypertrophic Cardiomyopathy (EVIDENCE-HCM). Circulation. 2018;137(10):1015-1023. https://doi.org/10.1161/CIRCULATIONAHA.117.030437
- Elliott PM, Sharma S, Varnava A, Poloniecki J, Rowland E, McKenna WJ. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999;33(6):1596-1601. https://doi.org/10.1016/S0735-1097(99)00056-X
- Spirito P, Autore C, Rapezzi C, et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119(13):1703-1710. https://doi.org/10.1161/CIRCULATIONAHA.108.798314
- Bos JM, Maron BJ, Ackerman MJ, et al. Role of family history of sudden death in risk stratification and prevention of sudden death with implantable defibrillators in hypertrophic cardiomyopathy. Am J Cardiol. 2010;106(10):1481-1486. https://doi.org/10.1016/J.AMJCARD.2010.06.077
- Dimitrow PP, Chojnowska L, Rudziński T, et al. Sudden death in hypertrophic cardiomyopathy: old risk factors re-assessed in a new model of maximalized follow-up. Eur Heart J. 2010;31(24):3084-3093. https://doi.org/10.1093/EURHEARTJ/EHQ308
- Spirito P, Bellone P, Harris KM, Bernabò P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342(24):1778-1785. https://doi.org/10.1056/NEJM200006153422403
- Autore C, Bernabò P, Barillà CS, Bruzzi P, Spirito P. The prognostic importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to the severity of symptoms. J Am Coll Cardiol. 2005;45(7):1076-1080. https://doi.org/10.1016/J.JACC.2004.12.067
- Elliott PM, Gimeno Blanes JR, Mahon NG, Poloniecki JD, McKenna WJ. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet. 2001;357(9254):420-424. https://doi.org/10.1016/S0140-6736(00)04005-8
- Harris KM, Spirito P, Maron MS, et al. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation. 2006;114(3):216-225. https://doi.org/10.1161/CIRCULATIONAHA.105.583500
- Rowin EJ, Maron BJ, Haas TS, et al. Hypertrophic Cardiomyopathy With Left Ventricular Apical Aneurysm: Implications for Risk Stratification and Management. J Am Coll Cardiol. 2017;69(7):761-773. https://doi.org/10.1016/J.JACC.2016.11.063
- Ichida M, Nishimura Y, Kario K. Clinical significance of left ventricular apical aneurysms in hypertrophic cardiomyopathy patients: the role of diagnostic electrocardiography. J Cardiol. 2014;64(4):265-272. https://doi.org/10.1016/J.JJCC.2014.02.011
- Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol. 2003;42(5):873-879. https://doi.org/10.1016/S0735-1097(03)00827-1
- Wang W, Lian Z, Rowin EJ, Maron BJ, Maron MS, Link MS. Prognostic Implications of Nonsustained Ventricular Tachycardia in High-Risk Patients With Hypertrophic Cardiomyopathy. Circ Arrhythm Electrophysiol. 2017;10(3). https://doi.org/10.1161/CIRCEP.116.004604
- Corona-Villalobos CP, Sorensen LL, Pozios I, et al. Left ventricular wall thickness in patients with hypertrophic cardiomyopathy: a comparison between cardiac magnetic resonance imaging and echocardiography. Int J Cardiovasc Imaging. 2016;32(6):945-954. https://doi.org/10.1007/S10554-016-0858-4
- Bois JP, Geske JB, Foley TA, Ommen SR, Pellikka PA. Comparison of Maximal Wall Thickness in Hypertrophic Cardiomyopathy Differs Between Magnetic Resonance Imaging and Transthoracic Echocardiography. Am J Cardiol. 2017;119(4):643-650. https://doi.org/10.1016/J.AMJCARD.2016.11.010
- Maron MS, Lesser JR, Maron BJ. Management implications of massive left ventricular hypertrophy in hypertrophic cardiomyopathy significantly underestimated by echocardiography but identified by cardiovascular magnetic resonance. Am J Cardiol. 2010;105(12):1842-1843. https://doi.org/10.1016/J.AMJCARD.2010.01.367
- Weng Z, Yao J, Chan RH, et al. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc Imaging. 2016;9(12):1392-1402. https://doi.org/10.1016/J.JCMG.2016.02.031
- Chan RH, Maron BJ, Olivotto I, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484-495. https://doi.org/10.1161/CIRCULATIONAHA.113.007094
- Mentias A, Raeisi-Giglou P, Smedira NG, et al. Late Gadolinium Enhancement in Patients With Hypertrophic Cardiomyopathy and Preserved Systolic Function. J Am Coll Cardiol. 2018;72(8):857-870. https://doi.org/10.1016/J.JACC.2018.05.060
- Ismail TF, Jabbour A, Gulati A, et al. Role of late gadolinium enhancement cardiovascular magnetic resonance in the risk stratification of hypertrophic cardiomyopathy. Heart. 2014;100(23):1851-1858. https://doi.org/10.1136/HEARTJNL-2013-305471
- O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35(30):2010-2020. https://doi.org/10.1093/EURHEARTJ/EHT439
- Binder J, Attenhofer Jost CH, Klarich KW, et al. Apical hypertrophic cardiomyopathy: prevalence and correlates of apical outpouching. J Am Soc Echocardiogr. 2011;24(7):775-781. https://doi.org/10.1016/J.ECHO.2011.03.002
- Rowin EJ, Maron BJ, Carrick RT, et al. Outcomes in Patients With Hypertrophic Cardiomyopathy and Left Ventricular Systolic Dysfunction. J Am Coll Cardiol. 2020;75(24):3033-3043. https://doi.org/10.1016/J.JACC.2020.04.045
- Marstrand P, Han L, Day SM, et al. Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry. Circulation. 2020;141(17):1371-1383. https://doi.org/10.1161/CIRCULATIONAHA.119.044366
- Chan RH, Maron BJ, Olivotto I, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484-495. https://doi.org/10.1161/CIRCULATIONAHA.113.007094
- Östman-Smith I, Wettrell G, Keeton B, et al. Age- and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur Heart J. 2008;29(9):1160-1167. https://doi.org/10.1093/EURHEARTJ/EHN122
- Miron A, Lafreniere-Roula M, Steve Fan CP, et al. A Validated Model for Sudden Cardiac Death Risk Prediction in Pediatric Hypertrophic Cardiomyopathy. Circulation. 2020;142(3):217-229. https://doi.org/10.1161/CIRCULATIONAHA.120.047235
- Norrish G, Ding T, Field E, et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol. 2019;4(9):918-927. https://doi.org/10.1001/JAMACARDIO.2019.2861
- Wells S, Rowin EJ, Bhatt V, Maron MS, Maron BJ. Association Between Race and Clinical Profile of Patients Referred for Hypertrophic Cardiomyopathy. Circulation. 2018;137(18):1973-1975. https://doi.org/10.1161/CIRCULATIONAHA.117.032838
- Norrish G, Cantarutti N, Pissaridou E, et al. Risk factors for sudden cardiac death in childhood hypertrophic cardiomyopathy: A systematic review and meta-analysis. Eur J Prev Cardiol. 2017;24(11):1220-1230. https://doi.org/10.1177/2047487317702519
- Maron BJ, Rowin EJ, Casey SA, et al. Hypertrophic Cardiomyopathy in Adulthood Associated With Low Cardiovascular Mortality With Contemporary Management Strategies. J Am Coll Cardiol. 2015;65(18):1915-1928. https://doi.org/10.1016/J.JACC.2015.02.061
- Maron BJ, Rowin EJ, Casey SA, et al. Risk stratification and outcome of patients with hypertrophic cardiomyopathy >=60 years of age. Circulation. 2013;127(5):585-593. https://doi.org/10.1161/CIRCULATIONAHA.112.136085
- Norrish G, Ding T, Field E, et al. A validation study of the European Society of Cardiology guidelines for risk stratification of sudden cardiac death in childhood hypertrophic cardiomyopathy. Europace. 2019;21(10):1559-1565. https://doi.org/10.1093/EUROPACE/EUZ118
- Maron BJ, Rowin EJ, Casey SA, et al. Hypertrophic Cardiomyopathy in Children, Adolescents, and Young Adults Associated With Low Cardiovascular Mortality With Contemporary Management Strategies. Circulation. 2016;133(1):62-73. https://doi.org/10.1161/CIRCULATIONAHA.115.017633
- Rowin EJ, Sridharan A, Madias C, et al. Prediction and Prevention of Sudden Death in Young Patients (. Am J Cardiol. 2020;128:75-83. https://doi.org/10.1016/J.AMJCARD.2020.04.042
- O’Mahony C, Tome-Esteban M, Lambiase PD, et al. A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/American Heart Association risk stratification and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart. 2013;99(8):534-541. https://doi.org/10.1136/HEARTJNL-2012-303271
- Vriesendorp PA, Schinkel AFL, van Cleemput J, et al. Implantable cardioverter-defibrillators in hypertrophic cardiomyopathy: patient outcomes, rate of appropriate and inappropriate interventions, and complications. 2013;166(3):496-502. https://doi.org/10.1016/J.AHJ.2013.06.009
- Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298(4):405-412. https://doi.org/10.1001/JAMA.298.4.405
- Balaji S, DiLorenzo MP, Fish FA, et al. Risk factors for lethal arrhythmic events in children and adolescents with hypertrophic cardiomyopathy and an implantable defibrillator: an international multicenter study. Heart Rhythm. 2019;16(10):1462-1467.
- Decker JA, Rossano JW, Smith EO, et al. Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol. 2009;54(3):250-254.
- Kamp AN, von Bergen NH, Henrikson CA, et al. Implanted defibrillators in young hypertrophic cardiomyopathy patients: a multicenter study. Pediatr Cardiol. 2013;34(7):1620-1627.
- Smith BM, Dorfman AL, Yu S, et al. Clinical significance of late gadolinium enhancement in patients< 20 years of age with hypertrophic cardiomyopathy. The American Journal of Cardiology. 2014;113(7):1234-1239.
- Raja AA, Farhad H, Valente AM, et al. Prevalence and progression of late gadolinium enhancement in children and adolescents with hypertrophic cardiomyopathy. Circulation. 2018;138(8):782-792. https://doi.org/10.1161/CIRCULATIONAHA.117.032966
- Maron BJ, Spirito P, Shen WK, et al. Implantable Cardioverter-Defibrillators and Prevention of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. JAMA. 2007;298(4):405-412. https://doi.org/10.1001/JAMA.298.4.405
- Vriesendorp PA, Schinkel AFL, van Cleemput J, et al. Implantable cardioverter-defibrillators in hypertrophic cardiomyopathy: Patient outcomes, rate of appropriate and inappropriate interventions, and complications. American Heart Journal. 2013;166(3):496-502. https://doi.org/10.1016/J.AHJ.2013.06.009
- Lampert R, Olshansky B, Heidbuchel H, et al. Safety of sports for athletes with implantable cardioverter-defibrillators: Results of a prospective, multinational registry. Circulation. 2013;127(20):2021-2030. https://doi.org/10.1161/CIRCULATIONAHA.112.000447
- Okamura H, Friedman PA, Inoue Y, et al. Single-Coil Defibrillator Leads Yield Satisfactory Defibrillation Safety Margin in Hypertrophic Cardiomyopathy. Circulation Journal. 2016;80(10):CJ-16-0428. https://doi.org/10.1253/CIRCJ.CJ-16-0428
- Killu AM, Park JY, Sara JD, et al. Cardiac resynchronization therapy in patients with end-stage hypertrophic cardiomyopathy. EP Europace. 2018;20(1):82-88. https://doi.org/10.1093/EUROPACE/EUW327
- Gu M, Jin H, Hua W, et al. Clinical outcome of cardiac resynchronization therapy in dilated-phase hypertrophic cardiomyopathy. Journal of Geriatric Cardiology : JGC. 2017;14(4):238. https://doi.org/10.11909/J.ISSN.1671-5411.2017.04.002
- Rogers DPS, Marazia S, Chow AW, et al. Effect of biventricular pacing on symptoms and cardiac remodelling in patients with end-stage hypertrophic cardiomyopathy. Eur J Heart Fail. 2008;10(5):507-513. https://doi.org/10.1016/J.EJHEART.2008.03.006
- Maron BJ, Spirito P, Ackerman MJ, et al. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61(14):1527-1535.
- Bettin M, Larbig R, Rath B, et al. Long-Term Experience With the Subcutaneous Implantable Cardioverter-Defibrillator in Teenagers and Young Adults. JACC Clin Electrophysiol. 2017;3(13):1499-1506. https://doi.org/10.1016/J.JACEP.2017.08.017
- Silvetti MS, Pazzano V, Verticelli L, et al. Subcutaneous implantable cardioverter-defibrillator: is it ready for use in children and young adults? A single-centre study. Europace. 2018;20(12):1966-1973. https://doi.org/10.1093/EUROPACE/EUY139
- Pettit SJ, McLean A, Colquhoun I, Connelly D, McLeod K. Clinical experience of subcutaneous and transvenous implantable cardioverter defibrillators in children and teenagers. Pacing Clin Electrophysiol. 2013;36(12):1532-1538. https://doi.org/10.1111/PACE.12233
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16). https://doi.org/10.1161/CIR.0B013E31829E8776
- Rowin EJ, Mohanty S, Madias C, Maron BJ, Maron MS. Benefit of Cardiac Resynchronization Therapy in End-Stage Nonobstructive Hypertrophic Cardiomyopathy. JACC Clin Electrophysiol. 2019;5(1):131-133. https://doi.org/10.1016/J.JACEP.2018.08.018
- Cappelli F, Morini S, Pieragnoli P, et al. Cardiac Resynchronization Therapy for End-Stage Hypertrophic Cardiomyopathy: The Need for Disease-Specific Criteria. J Am Coll Cardiol. 2018;71(4):464-466. https://doi.org/10.1016/J.JACC.2017.11.040
- Friedman PA, McClelland RL, Bamlet WR, et al. Dual-chamber versus single-chamber detection enhancements for implantable defibrillator rhythm diagnosis: the detect supraventricular tachycardia study. Circulation. 2006;113(25):2871-2879. https://doi.org/10.1161/CIRCULATIONAHA.105.594531
- Theuns DAMJ, Klootwijk APJ, Goedhart DM, Jordaens LJLM. Prevention of inappropriate therapy in implantable cardioverter-defibrillators: results of a prospective, randomized study of tachyarrhythmia detection algorithms. J Am Coll Cardiol. 2004;44(12):2362-2367. https://doi.org/10.1016/J.JACC.2004.09.039
- Kolb C, Sturmer M, Sick P, et al. Reduced risk for inappropriate implantable cardioverter-defibrillator shocks with dual-chamber therapy compared with single-chamber therapy: results of the randomized OPTION study. JACC Heart Fail. 2014;2(6):611-619. https://doi.org/10.1016/J.JCHF.2014.05.015
- Peterson PN, Greenlee RT, Go AS, et al. Comparison of Inappropriate Shocks and Other Health Outcomes Between Single- and Dual-Chamber Implantable Cardioverter-Defibrillators for Primary Prevention of Sudden Cardiac Death: Results From the Cardiovascular Research Network Longitudinal Study of Implantable Cardioverter-Defibrillators. J Am Heart Assoc. 2017;6(11). https://doi.org/10.1161/JAHA.117.006937
- Defaye P, Boveda S, Klug D, et al. Dual- vs. single-chamber defibrillators for primary prevention of sudden cardiac death: long-term follow-up of the Défibrillateur Automatique Implantable-Prévention Primaire registry. Europace. 2017;19(9):1478-1484. https://doi.org/10.1093/EUROPACE/EUW230
- Hu ZY, Zhang J, Xu ZT, et al. Efficiencies and Complications of Dual Chamber versus Single Chamber Implantable Cardioverter Defibrillators in Secondary Sudden Cardiac Death Prevention: A Meta-analysis. Heart Lung Circ. 2016;25(2):148-154. https://doi.org/10.1016/J.HLC.2015.07.008
- COHEN LS, BRAUNWALD E. Amelioration of angina pectoris in idiopathic hypertrophic subaortic stenosis with beta-adrenergic blockade. Circulation. 1967;35(5):847-851.
- Adelman AG, Shah PM, Gramiak R, Wigle ED. Long-term propranolol therapy in muscular subaortic stenosis. Heart. 1970;32(6):804-811.
- Stenson RE, Flamm Jr MD, Harrison DC, Hancock EW. Hypertrophic subaortic stenosis: clinical and hemodynamic effects of long-term propranolol therapy. Am J Cardiol. 1973;31(6):763-773.
- Bonow RO, Rosing DR, Bacharach SL, et al. Effects of verapamil on left ventricular systolic function and diastolic filling in patients with hypertrophic cardiomyopathy. Circulation. 1981;64(4):787-796. https://doi.org/10.1161/01.CIR.64.4.787
- TOSHIMA H, KOGA Y, NAGATA H, TOYOMASU K, ITAYA K ichi, MATOBA T. Comparable Effects of Oral Diltiazem and Verapamil in the Treatment of Hypertrophic Cardiomyopathy Double-blind Crossover Study. Jpn Heart J. 1986;27(5):701-715.
- Rosing DR, Kent KM, Maron BJ, Epstein SE. Verapamil Therapy: A New Approach to the Pharmacologic Treatment of Hypertrophic Cardiomyopathy II. Effects on Exercise Capacity and Symptomatic Status. Accessed May 16, 2022. http://ahajournals.org
- Sherrid M v, Barac I, McKenna WJ, et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45(8):1251-1258.
- Sherrid M v, Shetty A, Winson G, et al. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circulation: Heart Failure. 2013;6(4):694-702.
- Adler A, Fourey D, Weissler‐Snir A, et al. Safety of outpatient initiation of disopyramide for obstructive hypertrophic cardiomyopathy patients. J Am Heart Assoc. 2017;6(6):e005152.
- Maron BJ, Dearani JA, Ommen SR, et al. Low operative mortality achieved with surgical septal myectomy: role of dedicated hypertrophic cardiomyopathy centers in the management of dynamic subaortic obstruction. J Am Coll Cardiol. 2015;66(11):1307-1308.
- Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46(3):470-476.
- Braunwald E, Ebert PA. Hemodynamic alterations in idiopathic hypertrophic subaortic stenosis induced by sympathomimetic drugs∗. The American Journal of Cardiology. 1962;10(4):489-495. https://doi.org/10.1016/0002-9149(62)90373-9
- Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114(21):2232-2239.
- Kirk CR, Gibbs JL, Thomas R, Radley-Smith R, Qureshi SA. Cardiovascular collapse after verapamil in supraventricular tachycardia. Arch Dis Child. 1987;62(12):1265-1266.
- Moran AM, Colan SD. Verapamil therapy in infants with hypertrophic cardiomyopathy. Cardiol Young. 1998;8(3):310-319.
- Florian Rader. EXPLORER-LTE cohort of the MAVA-LTE extension study . In: Presented at: ACC April 3. ACC; 2022.
- Florian Rader. EXPLORER-HCM Presented at the American College of Cardiology Annual Scientific Session (ACC 2022). In: Presented at: ACC April 3. ; 2022.
- VALOR-HCM: Mavacamten Significantly Reduces Need For Surgical Intervention in Patients With Obstructive HCM – American College of Cardiology. Accessed May 5, 2022. https://www.acc.org/Latest-in-Cardiology/Articles/2022/03/31/20/23/Sat-930am-VALOR-HCM-acc-2022
- Rowin EJ, Maron BJ, Lesser JR, Rastegar H, Maron MS. Papillary muscle insertion directly into the anterior mitral leaflet in hypertrophic cardiomyopathy, its identification and cause of outflow obstruction by cardiac magnetic resonance imaging, and its surgical management. Am J Cardiol. 2013;111(11):1677-1679. https://doi.org/10.1016/J.AMJCARD.2013.01.340
- di Tommaso L, Stassano P, Mannacio V, et al. Asymmetric septal hypertrophy in patients with severe aortic stenosis: the usefulness of associated septal myectomy. J Thorac Cardiovasc Surg. 2013;145(1):171-175. https://doi.org/10.1016/J.JTCVS.2011.10.096
- Teo EP, Teoh JG, Hung J. Mitral valve and papillary muscle abnormalities in hypertrophic obstructive cardiomyopathy. Curr Opin Cardiol. 2015;30(5):475-482. https://doi.org/10.1097/HCO.0000000000000200
- Batzner A, Pfeiffer B, Neugebauer A, Aicha D, Blank C, Seggewiss H. Survival After Alcohol Septal Ablation in Patients With Hypertrophic Obstructive Cardiomyopathy. J Am Coll Cardiol. 2018;72(24):3087-3094. https://doi.org/10.1016/J.JACC.2018.09.064
- Nguyen A, Schaff H v., Hang D, et al. Surgical myectomy versus alcohol septal ablation for obstructive hypertrophic cardiomyopathy: A propensity score-matched cohort. J Thorac Cardiovasc Surg. 2019;157(1):306-315.e3. https://doi.org/10.1016/J.JTCVS.2018.08.062
- Kimmelstiel C, Zisa DC, Kuttab JS, et al. Guideline-Based Referral for Septal Reduction Therapy in Obstructive Hypertrophic Cardiomyopathy Is Associated With Excellent Clinical Outcomes. Circ Cardiovasc Interv. 2019;12(7). https://doi.org/10.1161/CIRCINTERVENTIONS.118.007673
- Mitra A, Ghosh RK, Bandyopadhyay D, Ghosh GC, Kalra A, Lavie CJ. Significance of Pulmonary Hypertension in Hypertrophic Cardiomyopathy. Curr Probl Cardiol. 2020;45(6). https://doi.org/10.1016/J.CPCARDIOL.2018.10.002
- Ong KC, Geske JB, Hebl VB, et al. Pulmonary hypertension is associated with worse survival in hypertrophic cardiomyopathy. European heart journal Cardiovascular Imaging. 2016;17(6):604-610. https://doi.org/10.1093/EHJCI/JEW024
- Desai MY, Bhonsale A, Patel P, et al. Exercise echocardiography in asymptomatic HCM: Exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC: Cardiovascular Imaging. 2014;7(1):26-36. https://doi.org/10.1016/J.JCMG.2013.08.010
- Nguyen A, Schaff H v., Nishimura RA, et al. Determinants of Reverse Remodeling of the Left Atrium After Transaortic Myectomy. Ann Thorac Surg. 2018;106(2):447-453. https://doi.org/10.1016/J.ATHORACSUR.2018.03.039
- Finocchiaro G, Haddad F, Kobayashi Y, et al. Impact of Septal Reduction on Left Atrial Size and Diastole in Hypertrophic Cardiomyopathy. Echocardiography. 2016;33(5):686-694. https://doi.org/10.1111/ECHO.13158
- Blackshear JL, Kusumoto H, Safford RE, et al. Usefulness of Von Willebrand Factor Activity Indexes to Predict Therapeutic Response in Hypertrophic Cardiomyopathy. Am J Cardiol. 2016;117(3):436-442. https://doi.org/10.1016/J.AMJCARD.2015.11.016
- Blackshear JL, Stark ME, Agnew RC, et al. Remission of recurrent gastrointestinal bleeding after septal reduction therapy in patients with hypertrophic obstructive cardiomyopathy-associated acquired von Willebrand syndrome. J Thromb Haemost. 2015;13(2):191-196. https://doi.org/10.1111/JTH.12780
- Desai MY, Smedira NG, Dhillon A, et al. Prediction of sudden death risk in obstructive hypertrophic cardiomyopathy: Potential for refinement of current criteria. J Thorac Cardiovasc Surg. 2018;156(2):750-759.e3. https://doi.org/10.1016/J.JTCVS.2018.03.150
- McLeod CJ, Ommen SR, Ackerman MJ, et al. Surgical septal myectomy decreases the risk for appropriate implantable cardioverter defibrillator discharge in obstructive hypertrophic cardiomyopathy. Eur Heart J. 2007;28(21):2583-2588. https://doi.org/10.1093/EURHEARTJ/EHM117
- Nishimura RA, Otto CM, Bonow RO, et al. 2017 AHA/ACC Focused Update of the 2014 AHA/ACC Guideline for the Management of Patients With Valvular Heart Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2017;135(25):e1159-e1195. https://doi.org/10.1161/CIR.0000000000000503
- Kim LK, Swaminathan R v., Looser P, et al. Hospital Volume Outcomes After Septal Myectomy and Alcohol Septal Ablation for Treatment of Obstructive Hypertrophic Cardiomyopathy: US Nationwide Inpatient Database, 2003-2011. JAMA Cardiol. 2016;1(3):324-332. https://doi.org/10.1001/JAMACARDIO.2016.0252
- Pelliccia F, Pasceri V, Limongelli G, … CAI journal of, 2017 undefined. Long-term outcome of nonobstructive versus obstructive hypertrophic cardiomyopathy: a systematic review and meta-analysis. Elsevier. Accessed May 12, 2022. https://www.sciencedirect.com/science/article/pii/S016752731730726X
- Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoraci. J Am Coll Cardiol. 2011;58(25):e212-e260. https://doi.org/10.1016/J.JACC.2011.06.011
- Yancy CW, Mariell Jessup C, Chair Biykem Bozkurt V, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol. 2017;70(6):776-803. https://doi.org/10.1016/J.JACC.2017.04.025
- Spoladore R, Maron MS, D’Amato R, Camici PG, Olivotto I. Pharmacological treatment options for hypertrophic cardiomyopathy: high time for evidence. European Heart Journal. 2012;33(14):1724-1733. https://doi.org/10.1093/EURHEARTJ/EHS150
- Axelsson A, Iversen K, Vejlstrup N, et al. Efficacy and safety of the angiotensin II receptor blocker losartan for hypertrophic cardiomyopathy: the INHERIT randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2015;3(2):123-131. https://doi.org/10.1016/S2213-8587(14)70241-4
- Nguyen A, Schaff H v., Nishimura RA, et al. Apical myectomy for patients with hypertrophic cardiomyopathy and advanced heart failure. The Journal of Thoracic and Cardiovascular Surgery. 2020;159(1):145-152. https://doi.org/10.1016/J.JTCVS.2019.03.088
- Bourmayan C, Razavi A, Fournier C, et al. Effect of propranolol on left ventricular relaxation in hypertrophic cardiomyopathy: An echographic study. American Heart Journal. 1985;109(6):1311-1316. https://doi.org/10.1016/0002-8703(85)90357-6
- Alvares RF, Goodwin JF. Non-invasive assessment of diastolic function in hypertrophic cardiomyopathy on and off beta adrenergic blocking drugs. Br Heart J. 1982;48(3):204-212. https://doi.org/10.1136/HRT.48.3.204
- Wilmshurst PT, Thompson DS, Juul SM, Jenkins BS, Webb-Peploe MM. Effects of verapamil on haemodynamic function and myocardial metabolism in patients with hypertrophic cardiomyopathy. Br Heart J. 1986;56(6):544-553. https://doi.org/10.1136/HRT.56.6.544
- Udelson JE, Bonow RO, O’Gara PT, et al. Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation. 1989;79(5):1052-1060. https://doi.org/10.1161/01.CIR.79.5.1052
- Pacileo G, de Cristofaro M, Russo MG, Sarubbi B, Pisacane C, Calabrò R. Hypertrophic cardiomyopathy in pediatric patients: effect of verapamil on regional and global left ventricular diastolic function. The Canadian Journal of Cardiology. 2000;16(2):146-152.
- Guttmann OP, Pavlou M, O’Mahony C, et al. Prediction of thrombo-embolic risk in patients with hypertrophic cardiomyopathy (HCM Risk-CVA). Eur J Heart Fail. 2015;17(8):837-845. https://doi.org/10.1002/EJHF.316
- Jung H, Yang PS, Jang E, et al. Effectiveness and Safety of Non-Vitamin K Antagonist Oral Anticoagulants in Patients With Atrial Fibrillation With Hypertrophic Cardiomyopathy: A Nationwide Cohort Study. Chest. 2019;155(2):354-363. https://doi.org/10.1016/J.CHEST.2018.11.009
- Noseworthy PA, Yao X, Shah ND, Gersh BJ. Stroke and Bleeding Risks in NOAC- and Warfarin-Treated Patients With Hypertrophic Cardiomyopathy and Atrial Fibrillation. J Am Coll Cardiol. 2016;67(25):3020-3021. https://doi.org/10.1016/J.JACC.2016.04.026
- Dominguez F, Climent V, Zorio E, et al. Direct oral anticoagulants in patients with hypertrophic cardiomyopathy and atrial fibrillation. Int J Cardiol. 2017;248:232-238. https://doi.org/10.1016/J.IJCARD.2017.08.010
- Page RL, Joglar JA, Caldwell MA, et al. 2015 ACC/AHA/HRS guideline for the management of adult patients with supraventricular tachycardia: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm. 2016;13(4):e136-e221. https://doi.org/10.1016/J.HRTHM.2015.09.019
- Wilke I, Witzel K, MÜnch J, et al. High Incidence of De Novo and Subclinical Atrial Fibrillation in Patients With Hypertrophic Cardiomyopathy and Cardiac Rhythm Management Device. J Cardiovasc Electrophysiol. 2016;27(7):779-784. https://doi.org/10.1111/JCE.12982
- Mahajan R, Perera T, Elliott AD, et al. Subclinical device-detected atrial fibrillation and stroke risk: a systematic review and meta-analysis. Eur Heart J. 2018;39(16):1407-1415. https://doi.org/10.1093/EURHEARTJ/EHX731
- Rowin EJ, Hausvater A, Link MS, et al. Clinical Profile and Consequences of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation. 2017;136(25):2420-2436. https://doi.org/10.1161/CIRCULATIONAHA.117.029267
- Tanel RE, Walsh EP, Lulu JA, Saul JP. Sotalol for refractory arrhythmias in pediatric and young adult patients: initial efficacy and long-term outcome. Am Heart J. 1995;130(4):791-797. https://doi.org/10.1016/0002-8703(95)90079-9
- Zhao DS, Shen Y, Zhang Q, et al. Outcomes of catheter ablation of atrial fibrillation in patients with hypertrophic cardiomyopathy: a systematic review and meta-analysis. Europace. 2016;18(4):508-520. https://doi.org/10.1093/EUROPACE/EUV339
- Lapenna E, Pozzoli A, de Bonis M, et al. Mid-term outcomes of concomitant surgical ablation of atrial fibrillation in patients undergoing cardiac surgery for hypertrophic cardiomyopathy†. Eur J Cardiothorac Surg. 2017;51(6):1112-1118. https://doi.org/10.1093/EJCTS/EZX017
- Guttmann OP, Rahman MS, O’Mahony C, Anastasakis A, Elliott PM. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: systematic review. Heart. 2014;100(6):465-472. https://doi.org/10.1136/HEARTJNL-2013-304276
- Noseworthy PA, Yao X, Shah ND, Gersh BJ. Stroke and Bleeding Risks in NOAC- and Warfarin-Treated Patients With Hypertrophic Cardiomyopathy and Atrial Fibrillation. J Am Coll Cardiol. 2016;67(25):3020-3021. https://doi.org/10.1016/J.JACC.2016.04.026
- Dominguez F, Climent V, Zorio E, et al. Direct oral anticoagulants in patients with hypertrophic cardiomyopathy and atrial fibrillation. Int J Cardiol. 2017;248:232-238. https://doi.org/10.1016/J.IJCARD.2017.08.010
- Maron BJ, Olivotto I, Bellone P, et al. Clinical profile of stroke in 900 patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39(2):301-307. https://doi.org/10.1016/S0735-1097(01)01727-2
- van Velzen HG, Theuns DAMJ, Yap SC, Michels M, Schinkel AFL. Incidence of Device-Detected Atrial Fibrillation and Long-Term Outcomes in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2017;119(1):100-105. https://doi.org/10.1016/J.AMJCARD.2016.08.092
- Wilke I, Witzel K, MÜnch J, et al. High Incidence of De Novo and Subclinical Atrial Fibrillation in Patients With Hypertrophic Cardiomyopathy and Cardiac Rhythm Management Device. J Cardiovasc Electrophysiol. 2016;27(7):779-784. https://doi.org/10.1111/JCE.12982
- Santangeli P, Biase L di, Themistoclakis S, et al. Catheter ablation of atrial fibrillation in hypertrophic cardiomyopathy: long-term outcomes and mechanisms of arrhythmia recurrence. Circ Arrhythm Electrophysiol. 2013;6(6):1089-1094. https://doi.org/10.1161/CIRCEP.113.000339
- Providencia R, Elliott P, Patel K, et al. Catheter ablation for atrial fibrillation in hypertrophic cardiomyopathy: a systematic review and meta-analysis. Heart. 2016;102(19):1533-1543. https://doi.org/10.1136/HEARTJNL-2016-309406
- Bogachev-Prokophiev A v., Afanasyev A v., Zheleznev SI, et al. Concomitant ablation for atrial fibrillation during septal myectomy in patients with hypertrophic obstructive cardiomyopathy. J Thorac Cardiovasc Surg. 2018;155(4):1536-1542.e2. https://doi.org/10.1016/J.JTCVS.2017.08.063
- Rowin EJ, Maron BJ, Abt P, et al. Impact of Advanced Therapies for Improving Survival to Heart Transplant in Patients with Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;121(8):986-996. https://doi.org/10.1016/J.AMJCARD.2017.12.044
- Rowin EJ, Maron BJ, Kiernan MS, et al. Advanced heart failure with preserved systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant. Circ Heart Fail. 2014;7(6):967-975. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001435
- Connolly SJ, Dorian P, Roberts RS, et al. Comparison of beta-blockers, amiodarone plus beta-blockers, or sotalol for prevention of shocks from implantable cardioverter defibrillators: the OPTIC Study: a randomized trial. JAMA. 2006;295(2):165-171. https://doi.org/10.1001/JAMA.295.2.165
- Santangeli P, Muser D, Maeda S, et al. Comparative effectiveness of antiarrhythmic drugs and catheter ablation for the prevention of recurrent ventricular tachycardia in patients with implantable cardioverter-defibrillators: A systematic review and meta-analysis of randomized controlled trials. Heart Rhythm. 2016;13(7):1552-1559. https://doi.org/10.1016/J.HRTHM.2016.03.004
- Baquero GA, Banchs JE, Depalma S, et al. Dofetilide reduces the frequency of ventricular arrhythmias and implantable cardioverter defibrillator therapies. J Cardiovasc Electrophysiol. 2012;23(3):296-301. https://doi.org/10.1111/J.1540-8167.2011.02183.X
- Gao D, van Herendael H, Alshengeiti L, et al. Mexiletine as an adjunctive therapy to amiodarone reduces the frequency of ventricular tachyarrhythmia events in patients with an implantable defibrillator. J Cardiovasc Pharmacol. 2013;62(2):199-204. https://doi.org/10.1097/FJC.0B013E31829651FE
- Link MS, Bockstall K, Weinstock J, et al. Ventricular Tachyarrhythmias in Patients With Hypertrophic Cardiomyopathy and Defibrillators: Triggers, Treatment, and Implications. J Cardiovasc Electrophysiol. 2017;28(5):531-537. https://doi.org/10.1111/JCE.13194
- Wilkoff BL, et al. 2015 HRS/EHRA/APHRS/SOLAECE expert consensus statement on optimal implantable cardioverter-defibrillator programming and testing. Europace. 2017;19(4):580. https://doi.org/10.1093/EUROPACE/EUW260
- Santangeli P, di Biase L, Lakkireddy D, et al. Radiofrequency catheter ablation of ventricular arrhythmias in patients with hypertrophic cardiomyopathy: safety and feasibility. Heart Rhythm. 2010;7(8):1036-1042. https://doi.org/10.1016/J.HRTHM.2010.05.022
- Igarashi M, Nogami A, Kurosaki K, et al. Radiofrequency Catheter Ablation of Ventricular Tachycardia in Patients With Hypertrophic Cardiomyopathy and Apical Aneurysm. JACC: Clinical Electrophysiology. 2018;4(3):339-350. https://doi.org/10.1016/J.JACEP.2017.12.020
- Dukkipati SR, D’Avila A, Soejima K, et al. Long-term outcomes of combined epicardial and endocardial ablation of monomorphic ventricular tachycardia related to hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol. 2011;4(2):185-194. https://doi.org/10.1161/CIRCEP.110.957290
- Borne RT, Varosy PD, Masoudi FA. Implantable cardioverter-defibrillator shocks: epidemiology, outcomes, and therapeutic approaches. JAMA Intern Med. 2013;173(10):859-865. https://doi.org/10.1001/JAMAINTERNMED.2013.428
- Mehra MR, Canter CE, Hannan MM, et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J Heart Lung Transplant. 2016;35(1):1-23. https://doi.org/10.1016/J.HEALUN.2015.10.023
- Raskin JS, Liu JJ, Abrao A, Holste K, Raslan AM, Balaji S. Minimally invasive posterior extrapleural thoracic sympathectomy in children with medically refractory arrhythmias. Heart Rhythm. 2016;13(7):1381-1385. https://doi.org/10.1016/J.HRTHM.2016.03.041
- Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342(6):365-373. https://doi.org/10.1056/NEJM200002103420601
- Nguyen A, Schaff H v. Electrical storms in patients with apical aneurysms and hypertrophic cardiomyopathy with midventricular obstruction: A case series. J Thorac Cardiovasc Surg. 2017;154(6):e101-e103. https://doi.org/10.1016/J.JTCVS.2017.06.002
- Hebl VB, Miranda WR, Ong KC, et al. The Natural History of Nonobstructive Hypertrophic Cardiomyopathy. Mayo Clin Proc. 2016;91(3):279-287. https://doi.org/10.1016/J.MAYOCP.2016.01.002
- Rowin EJ, Maron MS, Chan RH, et al. Interaction of Adverse Disease Related Pathways in Hypertrophic Cardiomyopathy. Am J Cardiol. 2017;120(12):2256-2264. https://doi.org/10.1016/J.AMJCARD.2017.08.048
- Melacini P, Basso C, Angelini A, et al. Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2010;31(17):2111-2123. https://doi.org/10.1093/EURHEARTJ/EHQ136
- Pasqualucci D, Fornaro A, Castelli G, et al. Clinical Spectrum, Therapeutic Options, and Outcome of Advanced Heart Failure in Hypertrophic Cardiomyopathy. Circ Heart Fail. 2015;8(6):1014-1021. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001843
- Coats CJ, Rantell K, Bartnik A, et al. Cardiopulmonary Exercise Testing and Prognosis in Hypertrophic Cardiomyopathy. Circ Heart Fail. 2015;8(6):1022-1031. https://doi.org/10.1161/CIRCHEARTFAILURE.114.002248
- Magrì D, Re F, Limongelli G, et al. Heart Failure Progression in Hypertrophic Cardiomyopathy – Possible Insights From Cardiopulmonary Exercise Testing. Circ J. 2016;80(10):2204-2211. https://doi.org/10.1253/CIRCJ.CJ-16-0432
- Kato TS, Takayama H, Yoshizawa S, et al. Cardiac transplantation in patients with hypertrophic cardiomyopathy. Am J Cardiol. 2012;110(4):568-574. https://doi.org/10.1016/J.AMJCARD.2012.04.030
- Topilsky Y, Pereira NL, Shah DK, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail. 2011;4(3):266-275. https://doi.org/10.1161/CIRCHEARTFAILURE.110.959288
- Killu AM, Park JY, Sara JD, et al. Cardiac resynchronization therapy in patients with end-stage hypertrophic cardiomyopathy. Europace. 2018;20(1):82-88. https://doi.org/10.1093/EUROPACE/EUW327
- Rogers DPS, Marazia S, Chow AW, et al. Effect of biventricular pacing on symptoms and cardiac remodelling in patients with end-stage hypertrophic cardiomyopathy. Eur J Heart Fail. 2008;10(5):507-513. https://doi.org/10.1016/J.EJHEART.2008.03.006
- Gu M, Jin H, Hua W, et al. Clinical outcome of cardiac resynchronization therapy in dilated-phase hypertrophic cardiomyopathy. J Geriatr Cardiol. 2017;14(4):238-244. https://doi.org/10.11909/J.ISSN.1671-5411.2017.04.002
- Cappelli F, Morini S, Pieragnoli P, et al. Cardiac Resynchronization Therapy for End-Stage Hypertrophic Cardiomyopathy: The Need for Disease-Specific Criteria. J Am Coll Cardiol. 2018;71(4):464-466. https://doi.org/10.1016/J.JACC.2017.11.040
- Musumeci MB, Russo D, Limite LR, et al. Long-Term Left Ventricular Remodeling of Patients With Hypertrophic Cardiomyopathy. Am J Cardiol. 2018;122(11):1924-1931. https://doi.org/10.1016/J.AMJCARD.2018.08.041
- Yancy CW, Jessup M, Bozkurt B, et al. 2016 ACC/AHA/HFSA Focused Update on New Pharmacological Therapy for Heart Failure: An Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. Circulation. 2016;134(13):e282-e293. https://doi.org/10.1161/CIR.0000000000000435
- Grupper A, Park SJ, Pereira NL, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: Improving outcomes for a lethal disease. J Heart Lung Transplant. 2015;34(8):1042-1049. https://doi.org/10.1016/J.HEALUN.2015.03.012
- Muthiah K, Phan J, Robson D, et al. Centrifugal continuous-flow left ventricular assist device in patients with hypertrophic cardiomyopathy: a case series. ASAIO J. 2013;59(2):183-187. https://doi.org/10.1097/MAT.0B013E318286018D
- Patel SR, Saeed O, Naftel D, et al. Outcomes of Restrictive and Hypertrophic Cardiomyopathies After LVAD: An INTERMACS Analysis. J Card Fail. 2017;23(12):859-867. https://doi.org/10.1016/J.CARDFAIL.2017.09.011
- Saberi S, Wheeler M, Bragg-Gresham J, et al. Effect of moderate-intensity exercise training on peak oxygen consumption in patients with hypertrophic cardiomyopathy: a randomized clinical trial. Jama. 2017;317(13):1349-1357.
- Maron BJ, Levine BD, Washington RL, Baggish AL, Kovacs RJ, Maron MS. Eligibility and Disqualification Recommendations for Competitive Athletes with Cardiovascular Abnormalities: Task Force 2: Preparticipation Screening for Cardiovascular Disease in Competitive Athletes: A Scientific Statement from the American Heart Association and American College of Cardiology. Circulation. 2015;132(22):e267-e272. https://doi.org/10.1161/CIR.0000000000000238
- Turkowski KL, Martijn Bos J, Ackerman NC, Rohatgi RK, Ackerman MJ. Return-to-play for athletes with genetic heart diseases. Circulation. 2018;137(10):1086-1088. https://doi.org/10.1161/CIRCULATIONAHA.117.031306
- US Department of Transportation FAAdministration. Medical Certification. Accessed June 25, 2022. https://www.faa.gov/licenses_certificates/medical_certification/
- Bateman BT. What’s New in Obstetric Anesthesia: a focus on maternal morbidity and mortality. Int J Obstet Anesth. 2019;37:68-72. https://doi.org/10.1016/J.IJOA.2018.09.004
- Sillesen M, Hjortdal V, Vejlstrup N, Sørensen K. Pregnancy with prosthetic heart valves – 30 years’ nationwide experience in Denmark. Eur J Cardiothorac Surg. 2011;40(2):448-454. https://doi.org/10.1016/J.EJCTS.2010.12.011
- Eleid MF, Konecny T, Orban M, et al. High prevalence of abnormal nocturnal oximetry in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54(19):1805-1809. https://doi.org/10.1016/J.JACC.2009.07.030
- Claes GRF, van Tienen FHJ, Lindsey P, et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur Heart J. 2016;37(23):1815-1822. https://doi.org/10.1093/EURHEARTJ/EHV522
- Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, et al. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J. 2018;39(34):3165-3241. https://doi.org/10.1093/EURHEARTJ/EHY340
- Fumagalli C, Maurizi N, Day SM, et al. Association of Obesity With Adverse Long-term Outcomes in Hypertrophic Cardiomyopathy. JAMA Cardiol. 2020;5(1):65-72. https://doi.org/10.1001/JAMACARDIO.2019.4268
- Smith JR, Medina-Inojosa JR, Layrisse V, Ommen SR, Olson TP. Predictors of Exercise Capacity in Patients with Hypertrophic Obstructive Cardiomyopathy. J Clin Med. 2018;7(11). https://doi.org/10.3390/JCM7110447
- Thaman R, Varnava A, Hamid MS, et al. Pregnancy related complications in women with hypertrophic cardiomyopathy. Heart. 2003;89(7):752-756. https://doi.org/10.1136/HEART.89.7.752
- Claes GRF, van Tienen FHJ, Lindsey P, et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur Heart J. 2016;37(23):1815-1822. https://doi.org/10.1093/EURHEARTJ/EHV522
- Wang S, Cui H, Song C, et al. Obstructive sleep apnea is associated with nonsustained ventricular tachycardia in patients with hypertrophic obstructive cardiomyopathy. Heart Rhythm. 2019;16(5):694-701. https://doi.org/10.1016/J.HRTHM.2018.12.017
- Miller CAS, Maron MS, Estes NAM, et al. Safety, Side Effects and Relative Efficacy of Medications for Rhythm Control of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Am J Cardiol. 2019;123(11):1859-1862. https://doi.org/10.1016/J.AMJCARD.2019.02.051
- Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Kardiologia Polska (Polish Heart Journal). 2014;72(11):1054-1126.
- Colan SD, Lipshultz SE, Lowe AM, et al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation. 2007;115(6):773-781. https://doi.org/10.1161/CIRCULATIONAHA.106.621185
- Nugent AW, Daubeney PEF, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639-1646. https://doi.org/10.1056/NEJMOA021737
- Limongelli G, Monda E, Tramonte S, et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int J Cardiol. 2020;299:186-191. https://doi.org/10.1016/J.IJCARD.2019.06.073
- Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes analysis of 1866 deaths in the united states, 1980-2006. Circulation. 2009;119(8):1085-1092. https://doi.org/10.1161/CIRCULATIONAHA.108.804617
- Wilkinson JD, Lowe AM, Salbert BA, et al. Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2012;164(3):442-448. https://doi.org/10.1016/J.AHJ.2012.04.018
- Lai WW, Cohen MS, Geva T, Mertens L (Eds), Colan SD. Normal echocardiographic values for cardiovascular structures, Appendix 1. Echocardiography in Pediatric and Congenital Heart Disease,. Wiley-Blackwell, West Sussex, UK. Published online August 5, 2009:p.765. https://doi.org/10.1002/9781444306309.FMATTER
- Shapiro LM, McKenna WJ. Distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: a two-dimensional echocardiographic study. J Am Coll Cardiol. 1983;2(3):437-444. https://doi.org/10.1016/S0735-1097(83)80269-1
- Moak JP, Leifer ES, Tripodi D, Mohiddin SA, Fananapazir L. Long-term follow-up of children and adolescents diagnosed with hypertrophic cardiomyopathy: risk factors for adverse arrhythmic events. Pediatr Cardiol. 2011;32(8):1096-1105. https://doi.org/10.1007/S00246-011-9967-Y
- Jhaveri S, Komarlu R, Worley S, Shahbah D, Gurumoorthi M, Zahka K. Left Atrial Strain and Function in Pediatric Hypertrophic Cardiomyopathy. J Am Soc Echocardiogr. 2021;34(9):996-1006. https://doi.org/10.1016/J.ECHO.2021.04.014
- Dallaire F, Slorach C, Hui W, et al. Reference values for pulse wave Doppler and tissue Doppler imaging in pediatric echocardiography. Circ Cardiovasc Imaging. 2015;8(2). https://doi.org/10.1161/CIRCIMAGING.114.002167
- Mozaffarian D, Caldwell JH. Right ventricular involvement in hypertrophic cardiomyopathy: a case report and literature review. Clin Cardiol. 2001;24(1):2-8. https://doi.org/10.1002/CLC.4960240102
- Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89(6):727-737. https://doi.org/10.1016/J.MAYOCP.2014.01.025
- Tadros R, Francis C, Xu X, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53(2):128-134. https://doi.org/10.1038/S41588-020-00762-2
- Ackerman MJ, VanDriest SL, Ommen SR, et al. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39(12):2042-2048. https://doi.org/10.1016/S0735-1097(02)01900-9
- Moolman JC, Corfield VA, Posen B, et al. Sudden death due to troponin T mutations. J Am Coll Cardiol. 1997;29(3):549-555. https://doi.org/10.1016/S0735-1097(96)00530-X
- Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336(11):195-202. https://doi.org/10.1056/NEJM199703133361107
- Spirito P, Maron BJ. Relation between extent of left ventricular hypertrophy and occurrence of sudden cardiac death in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1990;15(7):1521-1526. https://doi.org/10.1016/0735-1097(90)92820-R
- Suda K, Kohl T, Kovalchin JP, Silverman NH. Echocardiographic predictors of poor outcome in infants with hypertrophic cardiomyopathy. Am J Cardiol. 1997;80(5):595-600. https://doi.org/10.1016/S0002-9149(97)00428-1
- Nagueh SF, Mikati I, Kopelen HA, Middleton KJ, Quiñones MA, Zoghbi WA. Doppler estimation of left ventricular filling pressure in sinus tachycardia. A new application of tissue doppler imaging. Circulation. 1998;98(16):1644-1650. https://doi.org/10.1161/01.CIR.98.16.1644
- Arghami A, Dearani JA, Said SM, O’Leary PW, Schaff H v. Hypertrophic cardiomyopathy in children. Ann Cardiothorac Surg. 2017;6(4):376-385. https://doi.org/10.21037/ACS.2017.07.04
- Quality A for HR and. Strategy 6I: Shared Decision Making. In: The CAHPS Ambulatory Care Improvement Guide: Practical Strategies for Improving Patient Experience. Accessed May 10, 2022. https://www. ahrq.gov/cahps/quality-improvement/improvement-guide/6-strategiesfor-improving/communication/strategy6i-shared-decisionmaking.html.
- AHRQ Health Literacy Universal Precautions Toolkit | Agency for Healthcare Research and Quality. Accessed May 11, 2022. https://www.ahrq.gov/health-literacy/improve/precautions/index.html
- Greenfield S, Kaplan SH, Ware Jr JE, Yano EM, Frank HJ. Patients’ participation in medical care: Effects on blood sugar control and quality of life in diabetes. Journal of General Internal Medicine. 1988;3:448-457.
- Greenfield S, Kaplan S, Ware JE. Expanding patient involvement in care: Effects on patient outcomes. Annals of Internal Medicine. 1985;102(4):520-528. https://doi.org/10.7326/0003-4819-102-4-520
- Assessing the Effects of Physician-Patient Interactions on the Outcomes of Chronic Disease on JSTOR. Accessed May 11, 2022. https://www.jstor.org/stable/3765658
- Guadagnoli E, Ward P. Patient participation in decision-making. Social Science & Medicine. 1998;47(3):329-339. https://doi.org/10.1016/S0277-9536(98)00059-8


{kind=link}