

TS. PHẠM HỮU VĂN
Lời mở đầu
Đột tử một vấn đề lớn của y học. Chúng ta đã biết về một bản tổng hợp lâm sàng hàng đầu về các nguyên nhân gây đột tử do tim (SCD). Từ các khu vực địa lý khác nhau trên thế giới xuất hiện quan điểm lâm sàng khác nhau đối với vấn đề SCD và làm sáng tỏ các yếu tố dự đoán và ước tính tỷ lệ lưu hành trong di truyền trên toàn thế giới.
Đây là một vấn đề rất lớn của y học, trong chuyên đề rộng lớn này chúng tôi muốn trình bày mang tính chất nền tảng cùng các đồng nghiệp quan tâm để tiếp tục mở rộng nhận thức, nghiên cứu trong thực hành lâm sàng hàng ngày.
Trước hết chúng tôi muốn đề cập đến cơ sở di truyền cho các kênh khác nhau. Ở đây, chúng ta có thể tìm hiểu về các hội chứng rối loạn nhịp tim di truyền và các kênh tim được biết là có đột biến gen trong các gen mã hóa cho các kênh ion tim có thể phá vỡ sự cân bằng của dòng ion của điện thế hoạt động dẫn đến kiểu hình điện tâm đồ bất thường. Lĩnh vực bệnh lý tim mạch đang mở rộng nhanh chóng với sự phát hiện ra các gen mới liên quan đến rối loạn nhịp tim bao gồm hội chứng QT dài, hội chứng Brugada, hội chứng QT ngắn và nhịp nhanh thất đa hình liên quan đến catecholamine. Sự không đồng nhất tồn tại trong biểu hiện bệnh ở những bệnh nhân mang đột biến giống nhau. Chẩn đoán các cá nhân bị ảnh hưởng với liệu pháp kịp thời và phù hợp có thể ngăn ngừa các biến cố thảm khốc. Việc xác định các gen tiềm ẩn các bệnh này đã cho phép xác định các cá nhân không có triệu chứng có nguy cơ đột tử tim mang đột biến gen gây ra bệnh. Do đó di truyền đã được đưa vào hướng dẫn lâm sàng về đột tử do tim. Phần này chúng ta sẽ xem xét ngắn gọn về cơ sở di truyền, sinh lý bệnh, phân tầng nguy cơ và phương pháp quản lý của nhóm hội chứng này.
Ngoài ra, chúng ta đã biết một số lượng đáng kể các chuyên gia thể thao nổi tiếng đã chết trong hoặc ngay sau khi hoạt động thể thao không rõ nguyên nhân. Những trường hợp này có liên quan đến rung thất đột ngột và đột tử do tim ở những người trẻ có tim bình thường.
Tiếp theo chúng tôi sẽ cung cấp một bản tóm tắt lịch sử về sự tiến hóa của khái niệm tái cực sớm từ mô tả ban đầu đến các tài liệu mới nhất và một hướng dẫn để giúp các bác sĩ đánh giá các cá thể có mô hình điện tâm đồ phổ biến này.
Được biết, trong hơn 70 năm, tái cực sớm đã được coi là một biến thể bình thường phổ biến. Trong dân số nói chung, tỷ lệ phổ biến nằm trong khoảng từ 5 đến 13% và ở các vận động viên, xu hướng tăng được quan sát thấy từ 20 đến 90%. Tuy nhiên, từ nửa cuối thập niên 1990, ngày càng có nhiều báo cáo trường hợp, hàng loạt, nghiên cứu quan sát và hồi cứu báo cáo sự hiện diện của các mẫu điện tâm đồ khác nhau được cho là tái cực sớm có thể tạo ra một dấu hiệu tiềm tàng cho nguy cơ đột tử tăng lên ở các trường hợp, các nhân bình thường, tạo ra một bóng khuất về đặc thù ECG này.
Trong nhiều thế kỷ, sự tử vong bí ẩn của các vận động viên chuyên nghiệp nổi tiếng đã khiến giới truyền thông và bác sĩ chú ý, đặc biệt là vì các vận động viên quá cố trước đây đã đạt được kết quả thể thao phi thường mà không gặp vấn đề gì về tim. Không dễ để xác định dịch tễ học chính xác của SCD trong thể thao. Phần lớn phụ thuộc vào các tiêu chí đưa vào được lựa chọn để phân tích, độ tuổi của các vận động viên, mức độ thành tích thể thao, kinh nghiệm thể thao, loại hình thể thao và các yếu tố khác.
Đề cập đến dịch tễ học SCD trong Thể thao và ngừng tim khi hoạt động thể chất, báo cáo thông tin trên các tư liệu phổ biến ghi nhận 1.866 trường hợp đột tử và các trường hợp không tử vong được quan sát thấy trong 38 môn thể thao. Tỷ lệ mới mắc SCD cho thấy mức tăng đáng kể về mặt thống kê 6% mỗi năm. Ngoài ra, dữ liệu về dịch tễ học thể thao liên kết với SCD khá đa dạng, tùy thuộc vào truyền thống thể thao quốc gia, độ tuổi, giới tính và tiêu chí đưa vào nhóm (thể thao chuyên nghiệp, thể thao học đường, hoạt động thể dục nói chung). Trong một số trường hợp, có thể có được bệnh sử y tế của các nạn nhân hoặc dữ liệu về sự hiện diện của một số bệnh hoặc tình trạng cụ thể hoặc các triệu chứng tiềm ẩn trước giai đoạn gây tử vong. Điều này cho thấy ngay cả những than phiền về sức khỏe không đặc hiệu trong việc huấn luyện vận động viên thường xuyên phải được các bác sĩ, huấn luyện viên và chính các vận động viên thực hiện nghiêm túc, vì họ có thể báo trước biến cố đe dọa đến tính mạng. Một số điều kiện ở các vận động viên, thường được coi là đe dọa đến tính mạng, chẳng hạn như ngất, ngược lại, không phải lúc nào cũng liên quan đến nguy cơ tử vong đột ngột, mặc dù nguy cơ đó luôn phải được loại trừ trước tiên.
Một vấn đề không thể thiếu gồm đề cập đến các cuộc kiểm tra sàng lọc ở những người chơi thể thao và nó có giá trị lâm sàng thực hành. Các yếu tố huyết động và điện sinh lý khác nhau đáp ứng tốt để gắng sức ở tim khỏe mạnh. Một số thay đổi sinh lý để đáp ứng với tập luyện aerobic cường độ cao cấp tính bao gồm tăng đáng kể mức tiêu thụ ôxy của cơ xương và cung lượng tim. Thông tin về đột tử tim (SCD) của bất cứ ai, bất cứ nơi nào trên hành tinh này thực sự rất bi thảm, và nó có thể để lại những hậu quả xã hội và tâm lý phức tạp, đặc biệt là khi biến cố bất ngờ này xảy ra trong trường hợp không có bất kỳ triệu chứng và cảnh báo nào trước đó. Tuy nhiên, có sự tranh cãi chung giữa hầu hết mọi người về các vận động viên là biểu tượng của sức khỏe và sức mạnh. Tuy nhiên, tỷ lệ bi thảm của SCD ở các vận động viên trẻ đã vô hiệu hóa thực tế này và tạo ra sự chú ý rộng rãi của xã hội. Tỷ lệ mắc bệnh cao hơn của các vận động viên so với các đối tác không phải vận động viên. Do đó, SCD ở các vận động viên như một vấn đề sức khỏe toàn cầu lớn đòi hỏi sự chú ý của các nhà nghiên cứu và sự xem xét khoa học.
Phần tiếp theo sẽ thảo luận về kiến thức hiện tại về dịch tễ học và sinh lý học của bệnh nhịp nhanh thất ác tính ở bệnh nhân mắc bệnh tim bẩm sinh (CHD) và dữ liệu theo dòng thời gian lâm sàng của nhịp nhanh thất trong một nhóm bệnh nhân mắc nhiều khiếm khuyết bẩm sinh. Đáng chú ý, nhiều hội chứng di truyền hiếm gặp khác nhau có liên quan đến CHD đang được chú trọng, và mối liên quan giữa các nhát bóp thất sớm, nhịp nhanh thất tạm thời, dai dẳng và rung thất đang được thảo luận. Vấn đề này cho thấy nhịp tim nhanh thất xuất hiện trung bình ở tuổi 40 như thế nào và mức độ hiếm gặp của chúng ở những bệnh nhân chỉ bị nhịp nhanh thất tạm thời.
Bệnh nhân trưởng thành mắc bệnh tim bẩm sinh có nguy cơ tử vong do đột tử tim do rối loạn nhịp nhanh thất ác tính. Tỷ lệ mắc bệnh nhịp nhanh thất được báo cáo lên tới 30% và chúng chủ yếu được báo cáo ở những bệnh nhân bị tứ chứng Fallot và chuyển vị các đại động mạch. Những rối loạn nhịp tim này có thể được đi trước bằng nhịp nhanh thất tạm thời. Tại thời điểm này, 90% bệnh nhân CHD dự kiến sẽ sống sót đến tuổi trưởng thành ở các nước đã phát triển. Những cải thiện trong chăm sóc sức khỏe đã làm tăng đáng kể tuổi thọ của bệnh nhân CHD. Tuy nhiên, cũng có một nhược điểm: dân số CHD già và ngày càng tăng thường trải qua nhiều quá trình phẫu thuật trong suốt cuộc đời, cần được các chuyên gia chăm sóc y tế lâu dài và tăng nguy cơ rối loạn nhịp tim và SCD
Tiếp đến cần đưa ra những hiểu biết mới về phụ nữ và SCD ít gặp hơn. Phần này xem xét nguyên nhân căn bản của SCD ở phụ nữ và tóm tắt các yếu tố nguy cơ chính của SCD như là hướng dẫn để xác định bệnh nhân có nguy cơ cao trong điều trị dự phòng. Dự đoán và phòng ngừa SCD là một lĩnh vực điều tra tích cực nhưng các hướng dẫn hiện hành nêu rõ phụ nữ có tỷ lệ tử vong đột ngột thấp hơn so với nam giới. Trong 2/3 phụ nữ đột tử, đột tử do tim là biểu hiện lâm sàng đầu tiên của bệnh tim mạch vành. Phụ nữ sau mãn kinh có gánh nặng bệnh tim mạch lớn nhất trong đó có SCD. Nguyên nhân của SCD ở phụ nữ ít rõ ràng hơn, do phụ nữ được đại diện trong các nghiên cứu về SCD và trải nghiệm biến cố SCD ít hơn ở nam giới. Một số nghiên cứu cho thấy bệnh mạch vành có từ trước dự đoán ít hơn ở phụ nữ và các nguyên nhân khác có nhiều khả năng hơn. Ngoài ra, phụ nữ bị SCD ít có khả năng mắc bệnh động mạch vành tiềm ẩn (CAD) hơn nam giới điều đó cần xác định các yếu tố nguy cơ khác với CAD hoặc rối loạn chức năng tâm thu. Suy tim với chức năng tâm thu thất trái được bảo tồn, tăng tính kích thích giao cảm có thể được đánh giá bằng sự hấp thu meta-iodo-benzyl-guanidine, trầm cảm và / hoặc sử dụng thuốc chống trầm cảm là những yếu tố nguy cơ phổ biến của SCD ở phụ nữ.
Phần tiếp gồm đề cập đến tỷ lệ SCD và bệnh cơ tim phì đại và ước tính nguy cơ chính xác có thể cực kỳ khó khăn. Điều đó gần đây đã trở thành nguồn tranh luận giữa các chuyên gia châu Âu và Mỹ. Mặc dù yếu tố nguy cơ chính của SCD chắc chắn là ngừng tim đã được hồi sức trước đó, việc xác định các cá nhân có nguy cơ SCD cao trong phòng ngừa tiên phát vẫn còn là một vấn đề gây tranh cãi. Trong vài năm qua, nghiên cứu tim mạch đang tiến triển rất nhanh trong việc khám phá các yếu tố nguy cơ mới sửa đổi. Do đó, phình mỏm LV, đột biến đa gen sarcomere, xơ hóa cơ tim (myocardial fibrosis: MF) và giai đoạn cuối (ES) bệnh cơ tim phì đại (HCM) đang ngày càng trở nên quan trọng hơn khi là sự phán quyết cuối cùng một cách đặc biệt trong các trường hợp có nguy cơ trung gian. Sự cần thiết của một thỏa thuận quốc tế là một vấn đề hấp dẫn mà không thể trì hoãn trong tương lai.
Tiếp theo cần thảo luận về những hiểu biết mới về bệnh Chagas, nguyên nhân gây tử vong chính ở các nước Mỹ Latinh và nó đang trở thành một vấn đề thực sự đối với sức khỏe cộng đồng. Trong dân số Tây Ban Nha nguyên nhân phổ biến của SCD bao gồm bệnh động mạch vành (CAD), bệnh Chagas và bệnh cơ tim giãn nguyên phát. Sau một thế kỷ kể từ khi mô tả về căn bệnh này, Chagas vẫn đại diện cho một thách thức lớn đối với sức khỏe cộng đồng ở Mỹ Latinh. Trong những thập kỷ qua, một số biện pháp can thiệp bao gồm các cấp độ phòng ngừa tiên phát, thứ phát và cấp ba (tertiary) của bệnh Chagas đã được thử. Việc kiểm soát cả hai truyền qua vật trung gian và truyền máu trên cơ sở truyền T. cruzi (phòng ngừa tiên phát) dựa trên truyền qua vật trung gian và truyền máu đã thành công ở nhiều vùng lưu hành, nhưng việc phát hiện sớm và điều trị căn nguyên của các đối tượng không có triệu chứng chủ yếu được sử dụng. Đột tử do tim là nguyên nhân gây tử vong phổ biến nhất trong bệnh Chagas (55% – 65%), sau đó là suy tim sung huyết ở 25% – 30% và thuyên tắc não hoặc phổi trong 10% -15%.
Hầu hết các trường hợp SCD xảy ra ở những bệnh nhân mắc bệnh cơ tim biểu hiện từ 30 đến 50 tuổi. 20% bệnh nhân chết đột ngột không có các triệu chứng trước đó. SCD ở bệnh nhân chagas thường liên quan đến rối loạn nhịp tim, ít gặp hơn đối với các biến cố huyết khối (thuyên tắc phổi hoặc não lớn) hoặc hiếm khi vỡ phình LV. Rung tâm thất đã được báo cáo là nguyên nhân chính gây tử vong của SCD. Nguy cơ đột tử tim không giống nhau ở tất cả các bệnh nhân mắc bệnh Chagas mạn tính và đây là lý do tại sao một số tài liệu đã cố gắng xác định các yếu tố dự đoán. Một hệ thống tính điểm sẽ rất cần thiết để thiết lập các chiến lược phòng ngừa hiệu quả. Các bệnh nhân được xác định là có nguy cơ tử vong cao có thể nhận được lợi ích từ liệu pháp tích cực, gồm các nghiên cứu điện sinh lý bằng cách sử dụng ICD.
Tiếp theo đề cập đến các công nghệ mới và đang được nổi lên để phát hiện nguy cơ SCD. Trong số các phương thức hình ảnh mới nổi được đề xuất cho nghiên cứu bệnh tim cấu trúc, hình ảnh cộng hưởng từ tim (CMR) đóng vai trò quan trọng trong chẩn đoán IHD, đánh giá chẩn đoán và tiên lượng bệnh nhân SCD và gia đình nạn nhân SCD. CMR cho phép phân biệt IHD cấp tính và mãn tính thông qua đánh giá phù cơ tim bằng các chuỗi có trọng số T2, xác định xơ hóa cơ tim bằng phân tích ánh xạ T1 và tính toán khối lượng ngoại bào (Extracellular Volume: ECV) và đánh giá vùng nhồi máu bằng hình ảnh gia tăng gadolinium trễ (Late gadolinium enhancement: LGE).
Một số nghiên cứu nhằm khám phá giá trị tiên lượng gia tăng của định lượng sẹo cơ tim đã làm nổi bật tầm quan trọng của các “vùng xám” – vùng tăng cường tương phản trung gian có khả năng đại diện cho các vùng cơ tim còn sống hoặc không còn sống có khả năng gây loạn nhịp tim – được chứng minh là dự đoán mạnh mẽ độc lập cho khả năng tạo VT có thể chấp thuận., điều trị ICD phù hợp và tử suất. LVEF giảm là tiều chuẩn chính cơ bản chỉ định cầy ICD hiện nay cho dự phòng SCD tiên phát. Tuy nhiên, bất kỳ chiến lược nào cho dự phòng tiên phát dựa trên cơ sở LVEF đơn thuần đều có những hạn chế lớn.
LGE là một kỹ thuật được thiết lập tốt, cung cấp độ chính xác phân biệt (discriminative) của LGE tốt hơn LVEF cho phân tần nguy cơ rối loạn nhịp trên các cá thể và là giải pháp khả thi cho nhu cầu lâm sàng chưa được đáp ứng.
Phần tiếp theo trình bày về những hiểu biết hiếm hoi và khan hiếm về ung thư tim mạch. Hiện tại, những bệnh nhân sống sót sau ung thư đang đạt tỷ lệ sống sót lên tới 90% trong một số bệnh ung thư như ung thư vú và họ được điều trị bằng hóa trị cổ điển như anthracycline hoặc các liệu pháp nhắm mục tiêu mới như thuốc ức chế VEGF hoặc các hình thức khác bao gồm xạ trị và đang phải đối mặt với một mối đe dọa mới đối với cuộc sống của họ gây ra do các phương pháp này gây tổn hại cho sự phát triển của các bệnh lý tim mạch khác nhau đang tạo ra một thách thức mới về sự sống còn và một trong những mối đe dọa quan trọng là sự phát triển của thảm họa đột tử tim, đó là mối quan tâm chính của chúng ta trong phần này.
Chiếu xạ tim đến một liều đủ cao có thể làm tổn thương bất kỳ thành phần nào của tim, bao gồm màng ngoài tim, cơ tim, van tim, động mạch vành, mao mạch và hệ thống dẫn truyền. Ví dụ, viêm màng ngoài tim cấp có thể diễn ra nhanh chóng trong quá trình xạ trị đối với ngực. Điều này có thể dẫn đến tràn dịch màng ngoài tim, tamponade và xơ hóa màng ngoài tim (các thay đổi siêu âm tim) và dẫn đến tổn thương huyết động có thể gây ra đột tử tim. Bệnh nhân ung thư bị huyết khối gấp 2-3 lần so với bệnh nhân không ung thư và cao nhất với các tổn thương di căn và / hoặc các yếu tố nguy cơ hình thành và thường các bệnh nhân bị ảnh hưởng có tiên lượng xấu. Thường các thuyên tắc phổi (PE) lớn ở bệnh nhân ung thư không được chẩn đoán. Quản lý độc tính tim mạch đúng cách sẽ cho phép chúng ta tiếp tục điều trị ung thư, đồng thời ngăn ngừa hoặc giảm độc tính cho tim và điều này sẽ dẫn đến tỷ lệ sống cao hơn, kéo dài và an toàn hơn.
Phần cuối cùng liên quan đến khuyến cáo Hướng dẫn của ESC 2015 gần đây nhất về cấy ICD ở bệnh nhân suy tim có triệu chứng (NYHA Class II-III) và LVEF ≤ 35 ở bệnh nhân khi điều trị thuốc tối ưu ít nhất 1 tháng, với chỉ định Class I, mức độ bằng chứng A cho thiếu máu cục bộ (ít nhất 60 ngày sau khi bị nhồi máu cơ tim) và B cho bệnh nhân không bị thiếu máu cục bộ. Không nghi ngờ gì siêu âm tim là phương thức hình ảnh có sẵn cao nhất cho phân tầng nguy cơ SCD, đã đạt được tiến bộ đáng kể so với phương pháp truyền thống trong đó LVEF hai chiều được áp dụng cho mục đích này. Trong số các phát triển đáng chú ý nhất siêu âm tim ba chiều và chẩn đoán hình ảnh căng cơ tim đáng chú ý và có thể cải thiện đáng kể quá trình phân tầng nguy cơ hiện tại của chúng ta. Gần đây, sức căng (strain), phản ánh sự thay đổi chiều dài hoặc độ dày của sợi cơ tim, đã được đưa vào thực hành lâm sàng thông thường. Sức căng được đo bằng siêu âm tim theo dõi lốm đốm, dựa trên sự hiện diện của các dấu âm thanh tự nhiên trong hình ảnh siêu âm, hoạt động như một dấu vân tay cho mỗi phân đoạn cơ tim có thể được theo dõi trong chu kỳ tim và cho phép mô tả sự biến dạng. Tất cả các điều kiện lâm sàng được biết là có liên kết trực tiếp đến SCD được thảo luận ở đây với những hiểu biết mới trong thực hành lâm sàng hàng ngày.
Phần 1
CÁC HỘI CHỨNG RỐI LOẠN NHỊP KẾT HỢP VỚI ĐỘT TỬ TIM
TÓM TẮT
Đột tử (Sudden death: SD) được định nghĩa là tử vong tự nhiên không thể đoán trước của một cá nhân khỏe mạnh. Nguyên nhân chính của SD là do các biến đổi của tim, và do đó được gọi là đột tử do tim (Sudden cardiac death: SCD). Những tiến bộ gần đây trong khoa tim mạch đã phát triển các hướng dẫn mới trong chẩn đoán, điều trị cũng như phòng ngừa các bệnh liên quan đến SCD. Mặc dù thực tế này, việc xác định sớm các cá nhân có nguy cơ vẫn là một trong những thách thức chính hiện nay đối với các nhà tim mạch học. Một nguyên nhân chính của SCD là những biến đổi tố bẩm và các bệnh tim cấu trúc mặc dù một số lượng đáng kể không cho thấy bất kỳ khiếm khuyết tim nào trong quá trình khám nghiệm tử thi. Trong những trường hợp chưa được giải quyết này, bệnh kênh ion được coi là nguyên nhân tiềm năng đầu tiên của SD. Do tất cả các bệnh này có nguồn gốc di truyền, các thành viên gia đình có thể có nguy cơ, mặc dù không có triệu chứng. Thật không may, SCD thường là biểu hiện lâm sàng đầu tiên và duy nhất của một bệnh tim di truyền vẫn không bị phát hiện bằng các nghiên cứu lâm sàng thông thường. Trong một số trường hợp, hoạt động thể chất có thể là tác nhân kích hoạt SCD như là triệu chứng đầu tiên. Những tiến bộ công nghệ gần đây trong di truyền học đã cải thiện việc sử dụng xét nghiệm di truyền vào lĩnh vực SCD. Nó có thể giúp xác định nguyên nhân tử vong ở những bệnh nhân bị ảnh hưởng, trong các trường hợp sau khi chết mà không có chẩn đoán kết luận cũng như các thành viên gia đình không có triệu chứng có nguy cơ. Phần này tập trung vào những tiến bộ gần đây về rối loạn nhịp tim liên quan đến SCD.
MỞ ĐẦU
Đột tử (SD) được định nghĩa là một biến cố bất ngờ và tự nhiên xảy ra trong vòng một giờ đầu tiên kể từ khi xuất hiện các triệu chứng ở một người khỏe mạnh và trong đó khám nghiệm tử thi kỹ lưỡng không chứng minh được nguyên nhân tử vong (Basso et al., 2010). Do đó, nó đại diện cho một gánh nặng to lớn đối với gia đình, cộng đồng và chăm sóc sức khỏe, đặc biệt là khi xảy ra ở trẻ em. Biến đổi tim là nguyên nhân chính của SD (SCD). Ở quần thể dân số > 50 tuổi, gần 80% trường hợp SCD là hậu quả của bệnh tim mạch vành, trong khi ở dân số < 35 tuổi, SCD chủ yếu là do biến đổi di truyền (Oliva et al., 2010). Với bản chất di truyền của nó, có ý nghĩa quan trọng đối với các thành viên gia đình. Do đó, xét nghiệm di truyền có mục tiêu để tăng hiệu suất chẩn đoán, tạo điều kiện sàng lọc di truyền theo tầng của các thành viên gia đình với xét nghiệm tập trung hơn (Campuzano et al., 2014b). Một hạn chế lớn trong chẩn đoán lâm sàng của các gia đình là sự xâm nhập không đầy đủ và biểu hiện thay đổi, cản trở việc xác định tất cả người thân có nguy cơ mắc SCD (Giudicessi và Ackerman, 2013). Trong một số trường hợp, hoạt động thể chất có thể là tác nhân kích hoạt SCD như là triệu chứng đầu tiên. Tập trung vào các hội chứng di truyền, hai nhóm chính đã được đưa ra: bệnh lý kênh ion, trong đó chất nền gây rối loạn nhịp tim được tìm thấy trong sự bất thường của các tính chất điện của tim mà không có bất kỳ khiếm khuyết cấu trúc nào, và bệnh cơ tim, trong đó rối loạn nhịp tim có liên quan đến sự hiện diện của tim thay đổi cấu trúc tim (Wellens et al., 2014). Nghiên cứu di truyền đã chỉ ra các hội chứng di truyền liên quan đến SCD là do sự thay đổi bệnh lý trong gen mã hóa bốn loại protein của tế bào cơ: sarcomeric, cytoskeletal, desmosomal và các kênh ion hoặc protein liên quan. Trong khi chất nền cơ học là khác nhau ở cả hai nhóm, điểm kết thúc là một đặc điểm chung (Campuzano et al., 2014a). Trong phần này, chúng tôi sẽ tập trung vào cơ sở di truyền của rối loạn nhịp tim liên quan đến SCD.
BỆNH KÊNH ION
Như đã đề cập ở trên, thuật ngữ “bệnh kênh ion”, đề cập đến bệnh tim được đặc trưng bằng tim bình thường về cấu trúc dẫn đến rối loạn nhịp tim, ngất và SCD (Martin et al., 2013). Những bệnh rối loạn nhịp tim này được gây ra do các biến thể bệnh lý chủ yếu ở các gen mã hóa các kênh ion tim (chủ yếu là natri, kali và canxi) hoặc các protein liên quan. Nói chung, tùy thuộc vào kênh ion nào bị ảnh hưởng, các hội chứng khác nhau sẽ biểu hiện. Tuy nhiên, cùng một hội chứng có thể cho thấy một mức độ chồng chéo nhất định nếu các loại kênh khác nhau có thể bị ảnh hưởng.
Bảng 1. Các gene kết hợp với các bệnh kênh ion
| Kênh | Bệnh | Di truyền | GENE (ID) |
|
Natri |
LQT |
Nhiễm sắc thể thường trội | SCN5A (6331)
SCN4B (6330) SCN1B (6324) |
| BrS | Nhiễm sắc thể thường trội | SCN5A (6331)
GPD1L (23171) SCN1B (6324) SCN3B (55800) SCN2B (6327) SCN10A (6336) |
|
|
Natri được kết hợp |
LQT | Nhiễm sắc thể thường trội | CAV3 (859)
SNTA1 (6640) |
|
BrS |
Nhiễm sắc thể thường trội |
RANGRF (29098) SLMAP (7871)
PKP2 (5318) |
|
|
Kali |
LQT |
Nhiễm sắc thể thường trội |
KCNQ1 (3784)
KCNH2 (3757) KCNE1 (3753) KCNE2 (9992) KCNJ5 (3762) KCNJ2 (3759) |
| Nhiễm sắc thể thường lặn | KCNQ1 (3784)
KCNE1 (3753) |
||
| SQT |
Nhiễm sắc thể thường trội |
KCNH2 (3757)
KCNQ1 (3784) KCNJ2 (3759) |
|
|
BrS |
Nhiễm sắc thể thường trội |
ABCC9 (10060)
KCNE3 (10008) KCNJ8 (3764) HCN4 (10021) KCND3 (3752) |
|
| Trội liên quan giới tính | KCNE5 (23630) | ||
| CPVT | Nhiễm sắc thể thường trội | KCNJ2 (3759) | |
| Kali được kết hợp | LQT | Nhiễm sắc thể thường trội | AKAP9 (10142) |
|
Canxi |
BrS |
Nhiễm sắc thể thường trội |
CACNA1C (775) CACNB2 (785) CACNA2D1 (781) TRPM4 (54795) |
| SQT | Nhiễm sắc thể thường trội | CACNA2D1 (781) | |
| LQT | Nhiễm sắc thể thường trội | CACNA1C (775) RYR2 (6262) CALM1 (801) CALM2 (805) | |
| CPVT | Nhiễm sắc thể thường trội | RYR2 (6262) | |
| Nhiễm sắc thể thường lặn | CASQ2 (845) | ||
|
Canxi được kết hợp |
LQT | Nhiễm sắc thể thường trội | ANK2 (284) |
| Nhiễm sắc thể thường lặn | TRDN (10345) | ||
| CPVT | Nhiễm sắc thể thường trội
Nhiễm sắc thể thường trội |
CALM1 (801)
CALM2 (805) |
|
| Nhiễm sắc thể thường lặn | TRDN (10345) |
BrS: Hội chứng Brugada, CPVT: Nhịp nhanh thất đa hình liên quan với catecholamine, LQTS: Hội chứng QT dài. SQTS: Hội chứng QT ngắn.
Ngoài ra, nó còn được biết rõ sự tương tác của di truyền và môi trường như là một công cụ sửa đổi kiểu hình (Campuzano et al., 2010). Các kênh ion là glycoprotein được gắn trong màng tế bào cơ cho phép dòng ion vào và ra khỏi tế bào để điều chỉnh sự chênh lệc điện học. Dòng ion di chuyển điện tích và gây ra sự hình thành điện thế hoạt động của tim (dòng điện), được điều chỉnh bằng cách mở và đóng đồng bộ các kênh theo độ chênh điện thế. Những bệnh này thường ảnh hưởng đến việc tạo và truyền điện qua tế bào cơ, một bước quan trọng trong hoạt động cơ điện, đòi hỏi sự phối hợp của một số nguyên tố, bao gồm các kênh ion và protein cấu trúc, được đặt tên là phức hợp đại phân tử kênh ion (Martin et al., 2013). Sự phức tạp của quá trình này vẫn là một hạn chế lớn để hiểu các cơ chế gây rối loạn nhịp tim.
Hiện nay, các bệnh kênh chính là: hội chứng Brugada (BrS), hội chứng QT dài (LQTS), hội chứng QT ngắn (SQTS) và nhịp nhanh thất đa hình liên quan catecholamine (CPVT) (Priori và BlomstromLundqvist, 2015). Chúng ta sẽ tập trung vào 4 hội chứng rối loạn nhịp tim:
- Hội chứng Brugada
Hội chứng Brugada (BrS) là một thực thể hiếm gặp, được đặc trưng bằng sự chênh lên của đoạn ST type vòm (coved-type STsegment elevation) trong block nhánh bó phải không điển hình ở chuyển đạo V1 đến V3 của ECG, không có bệnh tim cấu trúc (Brugada và Brugada, 1992). Mẫu ECG có thể là cơ bản hoặc không liên tục, và nó có thể được bộc lộ trong quá trình test thuốc (thuốc chẹn kênh natri nhóm IC). Tỷ lệ hiện mắc được ước tính là 3-5 trên 10000 người mỗi năm và độ tuổi trung bình khởi phát các biến cố là khoảng 40 tuổi, nam giới bị ảnh hưởng là chính (75%). SCD liên quan đến BrS thường xảy ra trong khi ngủ, khi nghỉ ngơi hoặc khi tăng trương lực phế vị (Brugada et al., 2014). Bệnh nhân mắc BrS thường vẫn không có triệu chứng và các yếu tố điều biến như sốt, gắng sức hoặc thuốc (www.brugadadrugs.org), có thể đóng vai trò chính trong đặc tính động học của ECG. Mặc dù SCD trong hội chứng này không phổ biến trong khi gắng sức, khuyến cáo cho các vận động viên cạnh tranh với bất thường về tim mạch, khuyến cáo bệnh nhân BrS nên tránh tập thể dục cường độ cao. Việc quản lý bệnh nhân được chẩn đoán BrS là khó khăn vì thiếu liệu pháp y tế hiệu quả để ngăn ngừa rối loạn nhịp tim. Do đó, các kế hoạch phân tầng nguy cơ khá hạn chế vì ít được biết về biểu hiện lâm sàng và tiên lượng. Ở những bệnh nhân có chẩn đoán BrS và tiền sử ngất, SD được cứu sống, thở ngắc ngoải về ban đêm (nocturnal agonal breathing) hoặc co giật về đêm, được coi là một nhóm bệnh nhân có nguy cơ SCD cao hơn, và cấy ICD được chỉ định. Hiện tại, ICD là biện pháp phòng ngừa tốt nhất trong BrS. Tuy nhiên, cấy ICD khi có triệu chứng không phải là không có tranh cãi, đặc biệt là ở trẻ em (Sarquella-Brugada et al., 2016). Quyết định cấy ICD ở trẻ em không phải là một công việc dễ dàng vì đây là một liệu pháp suốt đời, không thể không bị biến chứng.
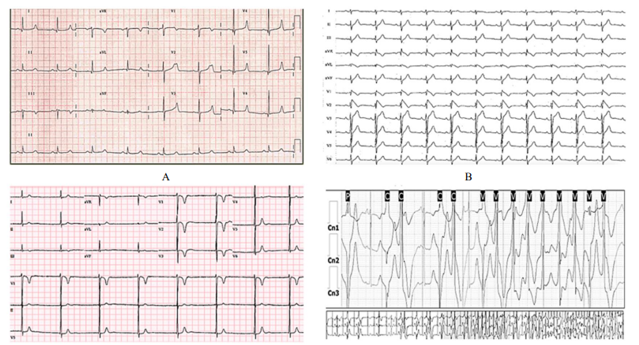
Hình 1. Điện tâm đồ cho thấy A. Hội chứng QT dài. B. Hội chứng Brugada. C. Hội chứng QT ngắn. D. Nhịp nhanh thất đa hình liên quan catecholamine.
Có dữ liệu hạn chế chứng minh lợi ích của các liệu pháp phòng ngừa khác nhưng nếu bệnh nhân cũng có biểu hiện rối loạn nhịp tim, Quinidine có thể được chỉ định khi bệnh nhân đã được cấy ICD, gây ra bão điện. BrS có thể liên kết rối loạn dẫn truyền AV và nhịp nhanh trên thất, vì vậy chúng ta đặt câu hỏi về sự có mặt của đánh trống ngực và cuối cùng điều trị các rối loạn nhịp tim này bằng triệt phá bằng năng lượng tần số radio qua catheter có thể được xem xét (Arbelo và Brugada, 2014).
Liên quan đến các nền tảng di truyền, cho đến nay hơn 250 biến thể gây bệnh đã được xác định trong 19 gen: [ABCC9, CACNA1C, CACNA2D1, CACNB2b, GPD1-L, HCN4, KCND3, KCNE3, KCNE5, KCNJ8, KCN, SCN3B, SCN5A, SLMAP và TRPM4] (Sarquella-Brugada và cộng sự., 2016). Một phân tích di truyền toàn diện chỉ xác định nguyên nhân gây bệnh ở 35% -40% gia đình và khoảng 25% – 30% trong số những bệnh nhân dương tính này có biến thể gây bệnh ở SCN5A (BrS type 1). Do đó, các hướng dẫn hiện tại chỉ đề xuất phân tích di truyền của gen này (Priori et al., 2015). Gen SCN5A mã hóa kênh natri tim Nav1.5 và các biến thể gây bệnh gây mất chức năng (Chen và cộng sự., 1998). Các biến thể gây bệnh khác đã được công bố trong các tiểu đơn vị beta làm thay đổi Nav1.5 (tăng hoặc giảm INa) và được mã hóa bằng SCN1B, SCN2B và SCN3B (Hu và cộng sự., 2009; Riuro và cộng sự., 2013; Watanabe và cộng sự., 2008). Gần đây, người ta đã công bố ngụ ý của SCN10A, một gen mã hóa kênh natri thần kinh Nav1.8 điều chỉnh biểu lộ Nav1.5 (Bezzina và cộng sự., 2013), nhưng một số nghiên cứu cần được thực hiện để làm rõ vai trò của gen này trong BrS. Ngoài ra, các biến thể gây bệnh trong gen GPD1-L làm giảm cả biểu hiện màng bề mặt và dòng natri vào bên trong (London cộng sự., 2007). Gần đây, gen RANGRF (protein MOG1) đã được báo cáo làm suy yếu việc trao đổi Nav1.5 đến màng (Kattygnarath cộng sự., 2011). Vào năm 2012, một biến thể gây bệnh đã được xác định trong SLMAP, một gen mã hóa protein liên quan đến màng sarcolemmal, được định vị tại các ống T và mạng lưới sarcoplasmic và điều chỉnh việc trao đổi nội bào của kênh hNav1.5 (Ishikawa cộng sự., 2012). Gần đây, người ta cũng đã báo cáo các biến thể gây bệnh trong PKP2 (protein plakophilin-2). Đây là một gen desmosomal và mối tương quan giữa sự mất biểu hiện của plakophilin-2 và Ina giảm (Cerrone và Delmar, 2014; Cerrone và cộng sự., 2014). Liên quan đến các kênh kali, bằng chứng đầu tiên cho thấy biến thể gây bệnh mới có chức năng gây bệnh trong KCND3 liên quan đến BrS đã được công bố vào năm 2011 (Giudicessi và cộng sự., 2011). Gen này mã hóa một thành phần của kênh kali, bị kiểm soát điện áp và nổi trội trong giai đoạn tái cực của điện thế hoạt động. Những thay đổi di truyền khác đã được báo cáo trong gen KCNE3 (protein MiRPS2) mã hóa một tiểu đơn vị điều tiết của kênh kali dưới đơn vị β của kênh Ito truyền kali ra ngoài (Delpon và cộng sự., 2008). Mặc dù BrS chủ yếu theo mô hình di truyền trội tự phát, một biến thể gây bệnh liên quan đến BrS đã được định vị trong gen KCNE1L (KCNE5) – X-link- (Ohno cộng sự., 2011). Gần đây, một gia đình BrS mang biến thể gây bệnh trong gen KCNJ8 cũng đã được báo cáo (Medeiros-Domingo và cộng sự, 2010). Ngoài ra, BrS cũng được liên kết với HCN4 mã hóa kênh 4 kali qua cổng nucleotide vòng được hoạt hóa phân cực quá mức (Ueda và cộng sự., 2009). Nó được biểu hiện ở nút xoang và các tế bào của hệ thống dẫn truyền tim, và mất chức năng của protein này có liên quan đến rối loạn chức năng nút xoang. Gen ABCC9 mã hóa thụ thể sulfonylurea tiểu đơn vị SUR2A. Các biến thể gây bệnh trong gen này gây ra sự tăng chức năng trong I (K-ATP) khi kết hợp với mất chức năng trong SCN5A và có thể có kiểu hình rối loạn nhịp tim nghiêm trọng (Hu và cộng sự., 2014). Cuối cùng, sự thay đổi di truyền trong các kênh canxi hoặc protein liên quan cũng đã được báo cáo trong các gia đình BrS. Các biến thể di truyền trong CACNA1C chịu trách nhiệm cho một đơn vị kênh canxi loại L bị khiếm khuyết. Điều đó gây ra mất chức năng kênh, nhưng được liên kết với khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Các biến thể di truyền khác trong CACNB2b dẫn đến cùng dấu vết ECG cho thấy sự kết hợp giữa BrS và khoảng QT ngắn hơn (Antzelevitch và cộng sự., 2007). Năm 2010, gen CACNA2D1 được liên kết với BrS (Burashnikov và cộng sự., 2010). Cuối cùng, các biến thể di truyền gây bệnh trong gen TRPM4 cũng đã được báo cáo. Gen này mã hóa protein melastatin điện thế thụ cảm thể tạm thời số 4, kênh cation không chọn lọc được canxi hoạt hóa, và là thành viên của gia đình lớn của các gen điện thế thụ thể tạm thời (Stallmeyer et al., 2012).
- Hội chứng QT dài
Hội chứng QT dài (Long QT syndrome: LQTS) là một bệnh kênh tim không đồng nhất hiếm gặp, đặc trưng bằng sự kéo dài khoảng QT trên ECG (QTc> 460 ms nữ và> 450 ms nam) (Amin và cộng sự., 2013). Phổ của bất thường ECG gây ra sự mất ổn định điện bao gồm sóng T chẻ đôi hoặc hai pha và sóng T thay đổi luân phiên. Biểu hiện của LQTS thay đổi từ bệnh nhân không có triệu chứng đến các cơn ngất do nhịp nhanh thất (Torsade de Pointes) trong trạng thái của một tim bình thường về cấu trúc. LQTS có tỷ lệ mắc 1/2500 cá thể, là nguyên nhân chính gây SCD ở những người trẻ tuổi (Mizusawa và cộng sự, 2014). Ở tất cả các bệnh nhân, việc sử dụng beta-blocker ở liều cao rất được khuyến khích vì việc điều trị này làm giảm nguy cơ SCD mặc dù không cung cấp sự bảo vệ đầy đủ. Liều được điều chỉnh theo sự dung nạp với các thuốc này (www.torades.org). Tuy nhiên, tranh cãi tồn tại liên quan đến hiệu quả của thuốc chẹn beta chọn lọc tim, chẳng hạn như atenolol. Cấy ghép ICD là bắt buộc đối với những bệnh nhân bị SCD được cứu sống và những người có nguy cơ rối loạn nhịp tim gây tử vong (Behere và cộng sự., 2014). LQTS có thể là bẩm sinh hoặc mắc phải, nguyên nhân cuối cùng thường liên quan đến thuốc và mất cân bằng điện giải (hạ kali máu, hạ canxi máu và hạ kali máu). Dạng bẩm sinh có liên quan đến sự biến đổi bệnh lý trong các kênh ion và / hoặc các protein liên quan. Về di truyền học, LQTS có thể theo mô hình di truyền trội hoặc lặn tự phát. Hiện tại, hơn 1000 biến thể gây bệnh đã được xác định trong 18 gen (AKAP9, ANK2, CACNA1C, CALM1, CALM2, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, RYR2, SC1, SC1, SC). Tất cả các gen này cùng nhau chịu trách nhiệm cho 80% 85% của tất cả các trường hợp LQT (Nakano và Shimizu, 2016). Gần đây, việc sắp xếp lại bên trong (xóa và sao chép lớn) có liên quan đến LQTS, cho thấy nguyên nhân gây bệnh có thể là do các biến thể số sao chép (copy number variants: CNV) ảnh hưởng đến bất kỳ trong số 18 gen này. Tỷ lệ phát hiện CNV trong các gia đình LQTS dường như khoảng 2% -11%. Các gen chính liên quan đến LQTS là KCNQ1 – type 1- (30% -35%), KCNH2 -type 2- (25% -30%) và SCN5A type 3- (5% -10%) chịu trách nhiệm cho 65% – 75% của tất cả các trường hợp LQTS. Các hướng dẫn gần đây khuyên cáo chỉ nên thử nghiệm 3 gen chính này trong các gia đình được chẩn đoán mắc LQTS, đặc biệt là nếu có SCD của một thành viên trẻ trong gia đình (Priori và cộng sự., 2015). Các biến thể gây bệnh trong gen KCNQ1 chịu trách nhiệm chính gây ra khoảng QT kéo dài. Protein này liên kết với protein được mã hóa bằng gen KCNE1 (minK) để tạo thành phức hợp chức năng Iks (Bianchi et al., 1999). Các biến thể gây bệnh trong SCN5A gây ra khiếm khuyết chức năng dựa trên sự bất hoạt không hoàn toàn của kênh, do đó cho phép tiếp tục xâm nhập các ion natri vào tế bào trong quá trình tái cực và dẫn đến chức năng được gia tăng. Một gen liên quan đến bệnh khác là KCNH2 mã hóa tiểu đơn vị alpha của phức hợp Ikr (Sanguinetti et al., 1996); tiểu đơn vị alpha được xác định bằng gen KCNE2. Phức hợp Ikr này là tác nhân quan trọng nhất của quá trình tái cực nhanh trong giai đoạn 3. Các biến thể gây bệnh trong KCNH2 dẫn đến mất chức năng trong kênh Ikr và chiếm 35% đến 45% các trường hợp hội chứng LQT. Trong KCNE2, các biến thể gây bệnh cũng dẫn đến mất chức năng kênh. Một gen khác có liên quan đến hội chứng QT dài là KCNJ2 mã hóa protein Ik1 (Kir2.1) (hội chứng Tawil Anderson) (Tsuboi và Antzelevitch, 2006). Một gen kali khác liên quan đến hội chứng LQT là KCNJ5, mã hóa cho kênh Kir3.4 (còn được gọi là GIRK4) (Yang và cộng sự., 2010). Nó tạo thành các kênh đẳng hiệu hoặc các kênh không đồng nhất chức năng với Kir3.x khác, các kênh chịu trách nhiệm cho dòng protein kênh G được điều chỉnh bên trong (IKACh) và chủ yếu được biểu hiện ở nút xoang nhĩ, nút xoang thất và nhĩ. Một protein liên quan đến kali với LQT là protein neo A-kinase 9 (AKAPs), là protein tạo giàn xác định khu vực protein kinase A (PKA) và các protein khác điều chỉnh PKA (phosphatase hoặc các kinase khác) (Chen và cộng sự., 2007). Nó được mã hóa bằng gen AKAP9. Gen natri khác là SCN4B mã hóa tiểu đơn vị beta (NaVb4) của kênh natri. Tiểu đơn vị NaVb4 gây ra thay đổi âm tính về điện thế phụ thuộc natri trong kênh kích hoạt (MedeirosDomingo et al., 2007). Gần đây, một biến thể bệnh lý đã được xác định trong gen SCN1B chịu trách nhiệm về hội chứng LQT. Nó mã hóa cho hai đồng phân tim của tiểu đơn vị Navβ1: Navβ1 isoform a và Navβ1 isoform b (Riuro et al., 2014). Tập trung vào các protein liên quan đến natri, CAV3 mã hóa cho caveolin-3, đây là protein chính hình thành nên các khoảng trống trong cơ tim và cơ xương (Vatta et al., 2006). Protein kết hợp với natri khác là α1-Syntrophin, được mã hóa bằng gen SNTA1, một thành phần của protein liên quan đến loạn sản có chứa nhiều thành phần tương tác protein (Wu et al., 2008). Nó cũng đã được báo cáo một số biến thể di truyền trong gen canxi. Các biến thể gây bệnh trong CACNA1C gây ra hội chứng LQT type 8 (Hội chứng Timothy) (Schwartz et al., 2006). Đây là loại hội chứng QT dài không phổ biến, nhưng nó có liên quan đến tỷ lệ tử vong cao nhất. Gần đây, một vài trường hợp hội chứng LQT đã được xác định ở hai gen: CALM1 và CALM2. Hai gen này cùng với mã hóa CALM3 cho protein peaceodulin và các sản phẩm của chúng có trình tự axit amin giống hệt nhau và cả ba gen được biểu hiện ở tâm thất trái của con người (Boczek et al., 2016; Reed et al., 2015). Calmodulin là một protein liên kết Ca + 2 đa chức năng cần thiết cho sự tải nạp tín hiệu Ca + 2 để tác động đến hoạt động của các kênh ion tim, kinase và các protein mục tiêu khác trong tim. Cuối cùng, hội chứng LQT cũng đã được liên kết với ANK2, một loại protein canxi liên quan (Mohler et al., 2003). Gen này mã hóa cho protein Ankyrin-B. Ankyrin là các protein bộ điều hợp liên kết các protein màng, chất vận chuyển và các phân tử kết dính tế bào với tế bào. Biến thể trong ANK2 gây ra sự trao đổi Na + / Ca + 2 và rối loạn chức năng Na + / K + ATPase tạo ra hậu khử cực sớm và trễ của tế nào (EAD và DADs) để đáp ứng với catecholamine
- Hội chứng QT ngắn
Hội chứng QT ngắn (SQTS) là một thực thể gây chết người rất hiếm được mô tả vào năm 2000 (Gussak và cộng sự., 2000). Nó được đặc trưng bằng khoảng QT ngắn (QTc <330 ms), với T cao nhọn và khoảng ngắn giữa đỉnh và kết thúc của sóng T trong ECG. Mức độ nghiêm trọng của các biểu hiện lâm sàng của SQT rất khác nhau, từ không triệu chứng đến ngất tái phát và SCD. Độ tuổi khởi phát các biểu hiện lâm sàng có thể rất trẻ với các báo cáo về các dạng ác tính gây ra cả SCD ở trẻ sơ sinh (Mazzanti et al., 2014). Ngày nay, không có biện pháp điều trị bằng thuốc nào hiệu quả đã được chứng minh để ngăn ngừa rối loạn nhịp tim và cấy ICD là biện pháp duy nhất cho các trường hợp nguy cơ cao. Quinidine đã được thử nghiệm như một phương pháp điều trị để cố gắng kéo dài QT. Rất ít dữ liệu được báo cáo liên quan đến thực hành gắng sức trong SQTS. Vì các căng thẳng giao cảm (stress adrenergic) không liên quan đến sự phát triển của rối loạn nhịp tim đe dọa tính mạng, cũng như không có môn thể thao cạnh tranh cũng như không có hoạt động thời gian trung bình được có phép đến tận khi có nhiều tư liệu hơn có thể (Ackerman et al., 2015)
Tập trung vào di truyền học, một số lượng hạn chế các biến thể di truyền gây bệnh đã thấy có liên quan với SQTS. Những thay đổi di truyền này đã được xác định ở 6 gen khác nhau (KCNQ1, KCNJ2, KCNH2, CACNA1C, CACNB2 và CACNA2D1), theo mô hình di truyền nhiễm sắc thể trội (Behere và Weindling, 2015). Điều này phải là một sự thâm nhập cao và một phân tích di truyền toàn diện xác định sự thay đổi di truyền ở gần 60% các trường hợp được chẩn đoán lâm sàng. Hội chứng QT ngắn type 1 có liên quan đến các biến thể di truyền trong KCNH2 gây ra sự kích hoạt nhanh dòng kali, với chức năng Ikr tăng cường và điện thế hoạt động thất ngắn lại (Brugada và cộng sự., 2004). Hội chứng QT ngắn type 2 có liên quan đến biến thể di truyền ở KCNQ1, tăng cường chức năng của kênh kali, dẫn đến rút ngắn điện thế hoạt động (Hong và cộng sự, 2005). Hội chứng QT ngắn type 3 gây ra do các biến thể di truyền trong KCNJ2, dẫn đến tăng tốc điện thế hoạt động giai đoạn 3 (Priori và cộng sự, 2005). Các biến thể trong gen canxi cũng đã được báo cáo. Hai trong số các gen này (CACNA1C, CACNB2) được liên kết với BrS và rút ngắn khoảng QT, như đã trình bày ở trên (Antzelevitch và cộng sự, 2007). Gen canxi thứ ba là CACNA2D1 và chỉ có 1 biến thể gây bệnh có liên quan đến SQTS (CM111612) (Templin và cộng sự, 2011).
- Nhịp nhanh thất đa hình liên quan đến Catecholamine
Nhịp tim nhanh thất đa hình liên quan đến catecholamine (Catecholaminergic polymorphic ventricular tachycardia: CPVT) là một thực thể tim hiếm gặp đặc trưng bằng nhịp nhanh thất đa hình hai chiều có đặc tính bằng rối loạn nhịp tim nghiêm trọng ở tim bình thường (Refaat và cộng sự, 2016). Hội chứng này có liên quan đến ECG bình thường khi nghỉ (đôi khi có nhịp tim chậm và sóng U), và chúng được kích hoạt bằng kích thích adrenergic và biểu hiện khi gắng sức, căng thẳng hoặc cảm xúc mạnh. Rối loạn nhịp này xảy ra chủ yếu ở trẻ em và thanh thiếu niên và ngày càng được công nhận là nguyên nhân gây SCD không giải thích được ở người trẻ tuổi, chủ yếu ở nam giới trẻ tuổi (30% ở độ tuổi 40) (Mohamed và cộng sự, 2007). Người ta nhận thấy biến cố này xảy ra ở thời thơ ấu (trước 10 tuổi) nhưng các nghiên cứu gần đây cho thấy biểu hiện đầu tiên có thể xảy ra từ giai đoạn sơ sinh đến 40 tuổi. Chẩn đoán CPVT có thể khó khăn đặc biệt ở trẻ nhỏ. Theo dõi ECG gắng sức và Holter 24 giờ có thể rất hữu ích để loại trừ nhịp tim nhanh hai chiều ở trẻ nhỏ, hoạt động thể chất vì CPVT không thể được chẩn đoán bằng ECG lúc nghỉ hoặc các nghiên cứu về tim mạch khác. Điều trị hàng đầu ở các bệnh nhân CPVT là thuốc chẹn beta, đã làm giảm đáng kể ngất và SCD. Hướng dẫn điều trị dựa trên dữ liệu hạn chế; tuy nhiên, ICD được chỉ định cho bệnh nhân SCD được cứu sống hoặc CPVT trong khi gắng sức và ở thanh thiếu niên có CPVT được kiểm soát không hoàn toàn mặc dù đã dùng thuốc liều cao (Imberti cộng sự, 2016). Phẫu thuật loại bỏ hạch giao cảm tim cũng đã được đề xuất để kiểm soát CPVT ở bệnh nhân trơ với betablockers, mặc dù các rối loạn nhịp giảm sau phẫu thuật này có thể là tạm thời hoặc khởi phát muộn. Thể thao trong rối loạn nhịp này được chống chỉ định, bao gồm cả những bệnh nhân được điều trị bằng thuốc chẹn beta (Ackerman và cộng sự, 2015).
Hiện tại, CPVT được gây ra bằng phân phối canxi nội bào bị suy yếu do gần 200 biến thể di truyền gây bệnh ở 5 gen khác nhau (RyR2, CASQ2, KCNJ2, CALM1 và TRDN). Tất cả các gen này cùng nhau giải thích khoảng 60% trường hợp CPVT. Gen chính liên quan đến CPVT là RyR2, chịu trách nhiệm cho gần 50% tất cả các trường hợp (Laitinen và cộng sự, 2001). Thụ thể ryanodine là một kênh canxi nội bào nằm trong mạng lưới sarcoplasmatic và được kích hoạt bằng dòng canxi nhỏ, do đó cho phép dòng canxi được lưu trữ, rất quan trọng trong việc kích hoạt co bóp cơ tim. Một gen khác liên quan đến CPVT là CASQ2 mã hóa cơ tim thành viên gia đình của protein gia đình trong lưới cơ tương của các tế bào cơ điều hòa nồng độ canxi (calsequestrin) hoạt động như lưu trữ canxi bên trong các tế bào cơ (Lahat et al., 2001). Hai protein liên quan đến canxi cũng đã được báo cáo trong các trường hợp CPVT, Calmodulin (CALM1) (Marsman và cộng sự, 2014) và Triadin (TRDN) (Roux-Buisson và cộng sự., 2012). Cho đến nay, chỉ có hai biến thể di truyền đã được liên kết với CPVT (CM128791 và CM128792) trong CALM1. Gần đây, một mối liên hệ tiềm tàng của gen CALM2 trong các đặc điểm lâm sàng chồng chéo của LQTS và CPVT đã được công bố mặc dù vẫn cần để làm rõ vai trò gây bệnh của chúng (Makita et al., 2014). Cuối cùng, Triadin (TRDN) là một protein màng nguyên phân có chứa một miền xuyên màng duy nhất, liên quan đến việc gắn Calributionestrin vào lưới cơ tương và cho phép cặp chức năng của nó với thụ thể Ryanodine điều chỉnh sự phóng thích canxi của cơ tương. Tập trung vào KCNJ2, một vài biến thể di truyền đã được liên kết với CPVT và nó đã được báo cáo là một biến thể (CM111211) ở một bệnh nhân cho thấy hội chứng Andersen-Tawil và giống với CPVT
KẾT LUẬN
Trong vài năm qua, tim mạch học đã được hưởng lợi nhờ cải thiện trong di truyền chủ yếu được áp dụng để chẩn đoán và phòng ngừa SCD. Tuy nhiên, SCD vẫn là một nguyên nhân quan trọng gây tử vong chủ yếu ở dân số trẻ. Mặc dù một số gen đã được báo cáo trong các bệnh kênh ion, phần lớn các gia đình được chẩn đoán lâm sàng vẫn không có nguyên nhân di truyền của bệnh được công nhận. Ngoài ra, việc xác định sớm các cá nhân có nguy cơ và phân tầng nguy cơ là những thách thức chính của các bệnh rối loạn nhịp tim liên quan đến SCD. Do đó, các nghiên cứu sâu hơn kết hợp với gia đình, bác sĩ lâm sàng và các nhà nghiên cứu cơ bản đòi hỏi làm rõ các điểm này và áp dụng điều trị phòng ngừa mang tính các thể hóa.
TÀI LIỆU THAM KHẢO
- Ackerman, M.J., Zipes, D.P., Kovacs, R.J., Maron, B.J., 2015. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 10: The Cardiac Channelopathies: A Scientific Statement From the American Heart Association and American College of Cardiology. Circulation 132, e326-329. Amin, A.S., Pinto, Y.M., Wilde, A.A., 2013. Long QT syndrome: beyond the causal mutation. J. Physiol. 591, 4125-4139.
- Antzelevitch, C., Pollevick, G.D., Cordeiro, J.M., Casis, O., Sanguinetti, M.C., Aizawa, Y., Guerchicoff, A., Pfeiffer, R., Oliva, A., Wollnik, B., Gelber, P., Bonaros, E.P., Jr., Burashnikov, E., Wu, Y., Sargent, J.D., Schickel, S., Oberheiden, R., Bhatia, A., Hsu, L.F., Haissaguerre, M., Schimpf, R., Borggrefe, M., Wolpert, C., 2007. Loss-offunction mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442-449.
- Arbelo, E., Brugada, J., 2014. Risk stratification and treatment of brugada syndrome. Curr. Cardiol. Rep. 16, 508. Basso, C., Carturan, E., Pilichou, K., Rizzo, S., Corrado, D., Thiene, G., 2010. Sudden cardiac death with normal heart Molecular autopsy. Cardiovasc. Pathol. Behere, S.P., Shubkin, C.D., Weindling, S.N., 2014. Recent advances in the understanding and management of long QT syndrome. Curr. Opin. Pediatr. 26, 727- 733.
- Behere, S.P., Weindling, S.N., 2015. Inherited arrhythmias: The cardiac channelopathies. Annals of pediatric cardiology 8, 210-220.
- Bezzina, C.R., Barc, J., Mizusawa, Y., Remme, C.A., Gourraud, J.B., Simonet, F., Verkerk, A.O., Schwartz, P.J., Crotti, L., Dagradi, F., Guicheney, P., Fressart, V., Leenhardt, A., Antzelevitch, C., Bartkowiak, S., Borggrefe, M., Schimpf, R., Schulze-Bahr, E., Zumhagen, S., Behr, E.R., Bastiaenen, R., Tfelt-Hansen, J., Olesen, M.S., Kaab, S., Beckmann, B.M., Weeke, P., Watanabe, H., Endo, N., Minamino, T., Horie, M., Ohno, S., Hasegawa, K., Makita, N., Nogami, A., Shimizu, W., Aiba, T., Froguel, P., Balkau, B., Lantieri, O., Torchio, M., Wiese, C., Weber, D., Wolswinkel, R., Coronel, R., Boukens, B.J., Bezieau, S., Charpentier, E., Chatel, S., Despres, A., Gros, F., Kyndt, F., Lecointe, S., Lindenbaum, P., Portero, V., Violleau, J., Gessler, M., Tan, H.L., Roden, D.M., Christoffels, V.M., Le Marec, H., Wilde, A.A., Probst, V., Schott, J.J., Dina, C., Redon, R., 2013. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 45, 1044-1049.
- Bianchi, L., Shen, Z., Dennis, A.T., Priori, S.G., Napolitano, C., Ronchetti, E., Bryskin, R., Schwartz, P.J., Brown, A.M., 1999. Cellular dysfunction of LQT5-minK mutants: abnormalities of IKs, IKr and trafficking in long QT syndrome. Hum. Mol. Genet. 8, 1499-1507.
- Boczek, N.J., Gomez-Hurtado, N., Ye, D., Calvert, M.L., Tester, D.J., Kryshtal, D.O., Hwang, H.S., Johnson, C.N., Chazin, W.J., Loporcaro, C.G., Shah, M., Papez, A.L., Lau, Y.R., Kanter, R., Knollmann, B.C., Ackerman, M.J., 2016. Spectrum and Prevalence of CALM1-, CALM2-, and CALM3-Encoded Calmodulin Variants in Long QT Syndrome and Functional Characterization of a Novel Long QT SyndromeAssociated Calmodulin Missense Variant, E141G. Circ. Cardiovasc. Genet. 9, 136- 146.
- Brugada, P., Brugada, J., 1992. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 20, 1391-1396.
- Brugada, R., Campuzano, O., Sarquella-Brugada, G., Brugada, J., Brugada, P., 2014. Brugada syndrome. Methodist DeBakey cardiovascular journal 10, 25-28.
- Brugada, R., Hong, K., Dumaine, R., Cordeiro, J., Gaita, F., Borggrefe, M., Menendez, T.M., Brugada, J., Pollevick, G.D., Wolpert, C., Burashnikov, E., Matsuo, K., Wu, Y.S., Guerchicoff, A., Bianchi, F., Giustetto, C., Schimpf, R., Brugada, P., Antzelevitch, C., 2004. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109, 30-35.
- Burashnikov, E., Pfeiffer, R., Barajas-Martinez, H., Delpon, E., Hu, D., Desai, M., Borggrefe, M., Haissaguerre, M., Kanter, R., Pollevick, G.D., Guerchicoff, A., Laino, R., Marieb, M., Nademanee, K., Nam, G.B., Robles, R., Schimpf, R., Stapleton, D.D., Viskin, S., Winters, S., Wolpert, C., Zimmern, S., Veltmann, C., Antzelevitch, C., 2010. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872-1882.
- Campuzano, O., Allegue, C., Brugada, R., 2014a. [Genetics of sudden unexplained death]. Medicina clinica 142, 265-269. Campuzano, O., Allegue, C., Partemi, S., Iglesias, A., Oliva, A., Brugada, R., 2014b. Negative autopsy and sudden cardiac death. International journal of legal medicine 128, 599-606.
- Campuzano, O., Beltran-Alvarez, P., Iglesias, A., Scornik, F., Perez, G., Brugada, R., 2010. Genetics and cardiac channelopathies. Genet. Med. 12, 260-267.
- Cerrone, M., Delmar, M., 2014. Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 24, 184-190.
- Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H., Novelli, V., Kim, C., Tirasawadichai, T., Judge, D.P., Rothenberg, E., Chen, H.S., Napolitano, C., Priori, S.G., Delmar, M., 2014. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092-1103.
- Chen, L., Marquardt, M.L., Tester, D.J., Sampson, K.J., Ackerman, M.J., Kass, R.S., 2007. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. US 104, 20990-20995.
- Chen, Q., Kirsch, G.E., Zhang, D., Brugada, R., Brugada, J., Brugada, P., Potenza, D., Moya, A., Borggrefe, M., Breithardt, G., Ortiz-Lopez, R., Wang, Z., Antzelevitch, C., O’Brien, R.E., Schulze-Bahr, E., Keating, M.T., Towbin, J.A., Wang, Q., 1998. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293-296.
- Delpon, E., Cordeiro, J.M., Nunez, L., Thomsen, P.E., Guerchicoff, A., Pollevick, G.D., Wu, Y., Kanters, J.K., Larsen, C.T., Hofman-Bang, J., Burashnikov, E., Christiansen, M., Antzelevitch, C., 2008. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209-218. Giudicessi, J.R., Ackerman, M.J., 2013. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Translational research: the journal of laboratory and clinical medicine 161, 1-14.
- Giudicessi, J.R., Ye, D., Tester, D.J., Crotti, L., Mugione, A., Nesterenko, V.V., Albertson, R.M., Antzelevitch, C., Schwartz, P.J., Ackerman, M.J., 2011. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024-1032.
- Gussak, I., Brugada, P., Brugada, J., Wright, R.S., Kopecky, S.L., Chaitman, B.R., Bjerregaard, P., 2000. Idiopathic short QT interval: a new clinical syndrome? Cardiology 94, 99-102.
- Hong, K., Piper, D.R., Diaz-Valdecantos, A., Brugada, J., Oliva, A., Burashnikov, E., Santos-de-Soto, J., Grueso-Montero, J., Diaz-Enfante, E., Brugada, P., Sachse, F., Sanguinetti, M.C., Brugada, R., 2005. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 68, 433-440.
- Hu, D., Barajas-Martinez, H., Burashnikov, E., Springer, M., Wu, Y., Varro, A., Pfeiffer, R., Koopmann, T.T., Cordeiro, J.M., Guerchicoff, A., Pollevick, G.D., Antzelevitch, C., 2009. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ. Cardiovasc. Genet. 2, 270-278.
- Hu, D., Barajas-Martinez, H., Terzic, A., Park, S., Pfeiffer, R., Burashnikov, E., Wu, Y., Borggrefe, M., Veltmann, C., Schimpf, R., Cai, J.J., Nam, G.B., Deshmukh, P., Scheinman, M., Preminger, M., Steinberg, J., Lopez-Izquierdo, A., Ponce-Balbuena, D., Wolpert, C., Haissaguerre, M., Sanchez-Chapula, J.A., Antzelevitch, C., 2014. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int. J. Cardiol. 171, 431-442.
- Imberti, J.F., Underwood, K., Mazzanti, A., Priori, S.G., 2016. Clinical Challenges in Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ. Ishikawa, T., Sato, A., Marcou, C.A., Tester, D.J., Ackerman, M.J., Crotti, L., Schwartz, P.J., On, Y.K., Park, J.E., Nakamura, K., Hiraoka, M., Nakazawa, K., Sakurada, H., Arimura, T., Makita, N., Kimura, A., 2012. A novel disease gene for Brugada syndrome: sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 5, 1098-1107.
- Kattygnarath, D., Maugenre, S., Neyroud, N., Balse, E., Ichai, C., Denjoy, I., Dilanian, G., Martins, R.P., Fressart, V., Berthet, M., Schott, J.J., Leenhardt, A., Probst, V., Le Marec, H., Hainque, B., Coulombe, A., Hatem, S.N., Guicheney, P., 2011. MOG1: a new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 4, 261-268.
- Lahat, H., Eldar, M., Levy-Nissenbaum, E., Bahan, T., Friedman, E., Khoury, A., Lorber, A., Kastner, D.L., Goldman, B., Pras, E., 2001. Autosomal recessive catecholamineor exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21.
- Circulation 103, 2822-2827. Laitinen, P.J., Brown, K.M., Piippo, K., Swan, H., Devaney, J.M., Brahmbhatt, B., Donarum, E.A., Marino, M., Tiso, N., Viitasalo, M., Toivonen, L., Stephan, D.A., Kontula, K., 2001. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485-490.
- London, B., Michalec, M., Mehdi, H., Zhu, X., Kerchner, L., Sanyal, S., Viswanathan, P.C., Pfahnl, A.E., Shang, L.L., Madhusudanan, M., Baty, C.J., Lagana, S., Aleong, R., Gutmann, R., Ackerman, M.J., McNamara, D.M., Weiss, R., Dudley, S.C., Jr., 2007. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 116, 2260-2268.
- Makita, N., Yagihara, N., Crotti, L., Johnson, C.N., Beckmann, B.M., Roh, M.S., Shigemizu, D., Lichtner, P., Ishikawa, T., Aiba, T., Homfray, T., Behr, E.R., Klug, D., Denjoy, I., Mastantuono, E., Theisen, D., Tsunoda, T., Satake, W., Toda, T., Nakagawa, H., Tsuji, Y., Tsuchiya, T., Yamamoto, H., Miyamoto, Y., Endo, N., Kimura, A., Ozaki, K., Motomura, H., Suda, K., Tanaka, T., Schwartz, P.J., Meitinger, T., Kaab, S., Guicheney, P., Shimizu, W., Bhuiyan, Z.A., Watanabe, H., Chazin, W.J., George, A.L., Jr., 2014. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 7, 466-474.
- Marsman, R.F., Barc, J., Beekman, L., Alders, M., Dooijes, D., van den Wijngaard, A., Ratbi, I., Sefiani, A., Bhuiyan, Z.A., Wilde, A.A., Bezzina, C.R., 2014. A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol. 63, 259-266.
- Martin, C.A., Huang, C.L., Matthews, G.D., 2013. The role of ion channelopathies in sudden cardiac death: implications for clinical practice. Ann. Med. 45, 364-374.
- Mazzanti, A., Kanthan, A., Monteforte, N., Memmi, M., Bloise, R., Novelli, V., Miceli, C., O’Rourke, S., Borio, G., Zienciuk-Krajka, A., Curcio, A., Surducan, A.E., Colombo, M., Napolitano, C., Priori, S.G., 2014. Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 63, 1300-1308.
- Medeiros-Domingo, A., Kaku, T., Tester, D.J., Iturralde-Torres, P., Itty, A., Ye, B., Valdivia, C., Ueda, K., Canizales-Quinteros, S., Tusie-Luna, M.T., Makielski, J.C., Ackerman, M.J., 2007. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 116, 134-142. Medeiros-Domingo, A., Tan, B.H., Crotti, L., Tester, D.J., Eckhardt, L., Cuoretti, A., Kroboth, S.L., Song, C., Zhou, Q., Kopp, D., Schwartz, P.J., Makielski, J.C., Ackerman, M.J., 2010. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 7, 1466-1471. Mizusawa, Y., Horie, M., Wilde, A.A., 2014. Genetic and clinical advances in congenital long QT syndrome. Circ. J. 78, 2827-2833.
- Mohamed, U., Napolitano, C., Priori, S.G., 2007. Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 18, 791-797.
- Mohler, P.J., Schott, J.J., Gramolini, A.O., Dilly, K.W., Guatimosim, S., duBell, W.H., Song, L.S., Haurogne, K., Kyndt, F., Ali, M.E., Rogers, T.B., Lederer, W.J., Escande, D., Le Marec, H., Bennett, V., 2003. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421, 634-639.
- Nakano, Y., Shimizu, W., 2016. Genetics of long-QT syndrome. J. Hum. Genet. 61, 51- 55. Ohno, S., Zankov, D.P., Ding, W.G., Itoh, H., Makiyama, T., Doi, T., Shizuta, S., Hattori, T., Miyamoto, A., Naiki, N., Hancox, J.C., Matsuura, H., Horie, M., 2011. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ. Arrhythm. Electrophysiol. 4, 352-361.
- Oliva, A., Brugada, R., D’Aloja, E., Boschi, I., Partemi, S., Brugada, J., Pascali, V.L., 2010. State of the Art in Forensic Investigation of Sudden Cardiac Death. Am. J. Forensic. Med. Pathol. Priori, S.G., Blomstrom-Lundqvist, C., 2015. 2015 European Society of Cardiology Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death summarized by co-chairs. Eur. Heart J. 36, 2757- 2759.
- Priori, S.G., Blomstrom-Lundqvist, C., Mazzanti, A., Blom, N., Borggrefe, M., Camm, J., Elliott, P.M., Fitzsimons, D., Hatala, R., Hindricks, G., Kirchhof, P., Kjeldsen, K., Kuck, K.H., Hernandez-Madrid, A., Nikolaou, N., Norekval, T.M., Spaulding, C., Van Veldhuisen, D.J., 2015. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC) Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace 17, 1601-1687.
- Priori, S.G., Pandit, S.V., Rivolta, I., Berenfeld, O., Ronchetti, E., Dhamoon, A., Napolitano, C., Anumonwo, J., di Barletta, M.R., Gudapakkam, S., Bosi, G., Stramba-Badiale, M., Jalife, J., 2005. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800-807.
- Reed, G.J., Boczek, N.J., Etheridge, S.P., Ackerman, M.J., 2015. CALM3 mutation associated with long QT syndrome. Heart Rhythm 12, 419-422. Refaat, M.M., Hassanieh, S., Scheinman, M., 2016. Catecholaminergic Polymorphic Ventricular Tachycardia. Cardiac electrophysiology clinics 8, 233-237.
- Riuro, H., Beltran-Alvarez, P., Tarradas, A., Selga, E., Campuzano, O., Verges, M., Pagans, S., Iglesias, A., Brugada, J., Brugada, P., Vazquez, F.M., Perez, G.J., Scornik, F.S., Brugada, R., 2013. A missense mutation in the sodium channel beta2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum. Mutat. 34, 961-966.
- Riuro, H., Campuzano, O., Arbelo, E., Iglesias, A., Batlle, M., Perez-Villa, F., Brugada, J., Perez, G.J., Scornik, F.S., Brugada, R., 2014. A missense mutation in the sodium channel beta1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 11, 1202-1209.
- Roux-Buisson, N., Cacheux, M., Fourest-Lieuvin, A., Fauconnier, J., Brocard, J., Denjoy, I., Durand, P., Guicheney, P., Kyndt, F., Leenhardt, A., Le Marec, H., Lucet, V., Mabo, P., Probst, V., Monnier, N., Ray, P.F., Santoni, E., Tremeaux, P., Lacampagne, A., Faure, J., Lunardi, J., Marty, I., 2012. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 21, 2759-2767.
- Sanguinetti, M.C., Curran, M.E., Spector, P.S., Keating, M.T., 1996. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc. Natl. Acad. Sci. US 93, 2208-2212.
- Sarquella-Brugada, G., Campuzano, O., Arbelo, E., Brugada, J., Brugada, R., 2016. Brugada syndrome: clinical and genetic findings. Genet. Med. 18, 3-12.
- Schwartz, P.J., Spazzolini, C., Crotti, L., Bathen, J., Amlie, J.P., Timothy, K., Shkolnikova, M., Berul, C.I., Bitner-Glindzicz, M., Toivonen, L., Horie, M., Schulze-Bahr, E., Denjoy, I., 2006. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation 113, 783-790.
- Stallmeyer, B., Zumhagen, S., Denjoy, I., Duthoit, G., Hebert, J.L., Ferrer, X., Maugenre, S., Schmitz, W., Kirchhefer, U., Schulze-Bahr, E., Guicheney, P., 2012. Mutational spectrum in the Ca(2+)–activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 33, 109-117.
- Templin, C., Ghadri, J.R., Rougier, J.S., Baumer, A., Kaplan, V., Albesa, M., Sticht, H., Rauch, A., Puleo, C., Hu, D., Barajas-Martinez, H., Antzelevitch, C., Luscher, T.F., Abriel, H., Duru, F., 2011. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 32, 1077-1088.
- Tsuboi, M., Antzelevitch, C., 2006. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm 3, 328-335.
- Ueda, K., Hirano, Y., Higashiuesato, Y., Aizawa, Y., Hayashi, T., Inagaki, N., Tana, T., Ohya, Y., Takishita, S., Muratani, H., Hiraoka, M., Kimura, A., 2009. Role of HCN4 channel in preventing ventricular arrhythmia. J. Hum. Genet. 54, 115-121.
- Vatta, M., Ackerman, M.J., Ye, B., Makielski, J.C., Ughanze, E.E., Taylor, E.W., Tester, D.J., Balijepalli, R.C., Foell, J.D., Li, Z., Kamp, T.J., Towbin, J.A., 2006. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation 114, 2104-2112.
- Watanabe, H., Koopmann, T.T., Le Scouarnec, S., Yang, T., Ingram, C.R., Schott, J.J., Demolombe, S., Probst, V., Anselme, F., Escande, D., Wiesfeld, A.C., Pfeufer, A., Kaab, S., Wichmann, H.E., Hasdemir, C., Aizawa, Y., Wilde, A.A., Roden, D.M., Bezzina, C.R., 2008. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260-2268.
- Wellens, H.J., Schwartz, P.J., Lindemans, F.W., Buxton, A.E., Goldberger, J.J., Hohnloser, S.H., Huikuri, H.V., Kaab, S., La Rovere, M.T., Malik, M., Myerburg, R.J., Simoons, M.L., Swedberg, K., Tijssen, J., Voors, A.A., Wilde, A.A., 2014. Risk stratification for sudden cardiac death: current status and challenges for the future. Eur. Heart J. 35, 1642-1651.
- Wu, G., Ai, T., Kim, J.J., Mohapatra, B., Xi, Y., Li, Z., Abbasi, S., Purevjav, E., Samani, K., Ackerman, M.J., Qi, M., Moss, A.J., Shimizu, W., Towbin, J.A., Cheng, J., Vatta, M., 2008. alpha-1-Syntrophin Mutation and the Long-QT Syndrome: A Disease of Sodium Channel Disruption. Circ. Arrhythm. Electrophysiol. 1, 193-201.
- Yang, Y., Liang, B., Liu, J., Li, J., Grunnet, M., Olesen, S.P., Rasmussen, H.B., Ellinor, P.T., Gao, L., Lin, X., Li, L., Wang, L., Xiao, J., Liu, Y., Zhang, S., Liang, D., Peng, L., Jespersen, T., Chen, Y.H., 2010. Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 86, 872-880.


{kind=link}