

Thuật ngữ y học “idiopathic” nằm trong cụm từ “idiopathic ventricular fibrillation” xuất phát từ nguồn gốc Hy Lạp: idios, hoặc “riêng của nó” (one’s own), và pathos, nghĩa là “đau khổ” hoặc “bệnh”.
TS Phạm Hữu Văn
Nghĩa đen là chứng bệnhgì đó như “bệnh của chính nó” (a disease of its own), hoặc một chứng bệnh không liên quan đến bất kỳ nguyên nhân cụ thể nào. Đây là chứng bệnhthường được thày thuốc chẩn đoán cuối cùng trên cơ sở loại trừ các các trạng thái bệnh kháccó xác địnhrõ nguyên nhân. Vì vậy thuật ngữ “idiopathic” chúng tôi đề nghị nên dịch ra tiếng Việt là “nguyên phát” hoặc “chưa rõ căn nguyên”. Chúng tôi biên soạn bài này với mục đích tổng quan lại nhóm bệnh có rung thất nhưng không có tổn thương tim thực thể, các chẩn đoán phân biệt để đi đến chẩn đoán VF nguyên phát, cũng như muốn thống nhất lại thuật ngữ liên quan đến nhóm bệnh này.
Rung thất nguyên phát (Idiopathic Ventricular Fibrillation- IVF) một bệnh ít gặp biểu hiện như ngất hoặc ngừng tim do nhịp nhanh thất (VT) đa hình nhanh hoặc rung thất (VF) khi không có bệnh tim thực thể. Trước đây người ta gọi VF nguyên phát là một bệnh chưa rõ căn nguyên. Mặc dù điều này là một thực tế cho phần lớn các ca ngày nay đã rõ có rung thất nguyên phát thực sự có biểu hiện bệnh lý kênh dưới dạng rung thất nguyên phát có tái cực sớm hoặc hội chứng QT ngắn bẩm sinh (SQTS) với khoảng QT không quá ngắn. Do thuật ngữ “nguyên phát” có nghĩa là “vắng mặt” VF có thể nhận biết căn nguyên đó là nền tảng chuẩn đoán loại trừ. Tuy nhiên các đặc tính điện sinh lý lâm sàng điển hình biểu hiện ở một số bệnh nhân thường cho phép chẩn đoán dương tính trực tiếp.
Lịch sử
Năm 1929, Dock đã xuất bản những gì có thể đại diện cho mô tả đầu tiên VF nguyên phát [1]. Báo cáo trường hợp này mô tả một nam giới 36 tuổi có một chuỗi cơn ngất do VF được chứng minh bằng tư liệu là nguyên phát. Bệnh tim thực thể đã được loại trừ phù hợp với các công nghệ có sẵn. Các trường hợp tương tự đã được thông báo tiếp theo và năm 1987 Belhassen xuất bản một loạt đầu tiên các VF nguyên phát [2], nhấn mạnh tầm quan trọng của việc đánh giá điện sinh lý bằng kích thích thất có chương trình và hiệu quả cao của điều trị quinidine để ngăn ngừa VF có thể được thúc đẩy và tự phát [2].
Năm 1990, người ta đã xuất bản bản nhận xét một cách hệ thống đầu tiên về rung thất nguyên phát (idiopathic VF) [3], gồm số liệu 54 trường hợp đã được xuất bản. Các đặc tính điển hình của VF nguyên phát, gồm khởi đầu các triệu chứng trong giai đoạn trưởng thành ở cả hai giới, tỷ lệ mắc các cơn bão loạn nhịp (với chuỗi các cơn VF) tương đối cao, tần số VF được tạo ra bằng kích thích thất có chương trình và đáp ứng tốt với điều trị quinidine, đã được tóm tắt lần đầu tiên trong bản nhận xét này.
Kiểu khởi phát các rối loạn nhịp tự nhiên trong VF nguyên phát, cụ thể là sự khởi kích VT/VF đa hình nhanh bằng các ngoại tâm thu thất đơn với các khoảng ghép rất ngăn (R-on-T), đã được Leenhardt và Coumel [4] chứng minh từ các báo cáo đầu tiên, như là biến thể có khoảng ghép ngắn trong xoắn đỉnh (torsade de pointes) và người mô tả chi tiết vào năm 1997 [5]. Sáu năm sau, Haissaguerre đã chứng minh các ngoại tâm thu được ghép ngắn khởi kích VF trong bệnh này là những nhắt bóp ngoại vị rất sớm có nguồn gốc từ các sợi Purkinje [6].
Chẩn đoán phân biệt của VF nguyên phát cũng đã được phát triển trong những năm gần đây. Khi người ta trình bày đầu tiên chủ đề này vào năm 1990 [3], chẩn đoán phân biệt gồm (ngoài các dạng tính tế của bệnh tim thực thể) các rối loạn nhịp sau: hội chứng QT kéo dài (mô tả năm 1957) [7-9] VT đa hình nhạy cảm với catecholamine (CPVT) (mô tả năm 1995) [10], và hội chứng tử vong đột ngộtvề ban đêm ở Đông Nam Á (được biết đến từ năm 1960) [11]. Tuy nhiên, vào năm 1992, anh em Brugada đã mô tả các bệnh nhân bị VF nguyên phát ở phương diện khác có biểu hiện điện tâm đồ đặc biệt cho thấy có block nhánh phải có đoạn ST chênh lên cố định ở các chuyển đạo trước tim phải [12]. Không lâu sau đó đã có bằng chứng > 20% bệnh nhân được cho có VF nguyên phát-ngày nay chúng ta gọi đó là “hội chứng Brugada” [13]. Hơn nữa, vào năm 1997, rõ ràng là “hội chứng đột tử về ban đêm ở Đông Nam Á” là một biểu hiện “đặc hữu” của hội chứng Brugada ở Châu Á [14]. Sau đó, vào năm 2000, hội chứng QT ngắn bẩm sinh đã được mô tả [15, 16], khi những bệnh nhân như vậy có các rối loạn nhịp thất có thể tạo ra cũng như tự phát [16] [17], không thể phân biệt được với bệnh nhân VF nguyên phát;người ta đã đề xuất vào năm 2004 VF nguyên phát có thể là “hội chứng QT ngắn với khoảng QT không quá ngắn” (các khoảng QTc trong khoảng 340-360 ms) [18]. Cuối cùng, năm 2008, Haissaguerre, Nam [20] và một nhóm nghiên cứu khác [21] phát hiệnthấy “các mẫu tái cực sớm” (sự kết hợp của sóng J và đoạn ST từ lâuđược tin là mẫu điện tâm đồ hoàn toàn lành tính)liên quan chặt chẽ với bệnh sử VF nguyên phát, tiếp tục ủng hộ cho khái niệm “Hội chứng sóng J” [22].
Bệnh căn
Thường xảy ra với các bệnh ban đầu được gọi là “vô căn”, nó nhiều khả năng như “VF nguyên phát” không đại diện cho một thực thể đơn lẻ mà đại diện cho các bệnh khác nhau với các đặc điểm điện tâm đồ tương tự, gồm các bệnh kênh (channelopathies) khác nhau. Ví dụ, một số trường hợp “VF nguyên phát” thực tế có thể có SQTS bẩm sinh. Mặc dù SQTS ban đầu được xác định là hội chứng rối loạn nhịp tim với QTc cơ bản < 300 ms [15, 16, 23], rõ ràng là không có giá trị QTc duy nhất nào khác biệt với tất cả các bệnh nhân khỏe mạnh so với tất cả các bệnh nhân có SQTS [24]. Trên thực tế, những người mang các đột biến SQTS với các khoảng QTc với thời gian 362 ms (được xem là trong phạm vi thấp của bình thường) [24] đã được mô tả rõ ràng [25]. Đồng thời, người ta đã chỉ ra những bệnh nhân VF nguyên phát (đặc biệt là nam giới) có khoảng QT ngắn hơn các nhóm chứng được tính theo tuổi (thường trong khoảng 360-370 ms) và những người khác cho thấy những bệnh nhân VF nguyên phát có nhịp tim bình thường ở mức cơ bản nhưng kéo dài QT không đạt trong quá trình nhịp tim chậm [26, 27], cho thấy VF nguyên phát có thể biểu hiện một chuỗi liên tục [24]. Thật thú vị, trong thân tộc rất lớn (liên quan đến ba gia đình khác biệt rõ ràng với đột biến người thế hệ đầu) của VF nguyên phát ở Hà Lan, những người mang haplotype nguy cơ có điện tâm đồ đã được xác định “rất bình thường”, và những người mang bệnh có các khoảng QT không có sự khác biệt với những người không mang bệnh [28]. Tuy nhiên, QTc trung bình của những người mang haplotype nguy cơ (395 ms) sẽ giảm ngắn hơn 10 bách phân vị của QTc của quần thể khỏe mạnh [29]. Ngoài ra, rối loạn di truyền nền được giả định (vượt quá mức DPP6) sẽ được cho là tăng dòng tạm thời đi ra ngoài (Ito), rút ngắn điện thế hoạt động ở một số khu vực hơn những người khác, một lần nữa hướng tới “giả thuyết QT ngắn”. Mặt khác, các trường hợp VF nguyên phát khác dường như có các sóng J được ghi nhận hơn là khoảng QT ngắn [19, 21]. Các mô hình thí nghiệm [22] và các báo cáo không thực sự cần thiết của các bệnh nhân VF nguyên phát có các sóng J được nghiên cứu bằng các phương pháp hình ảnh học không xâm lấn mới [30] gợi ý các gradient tái cực nhanh (thẳng đứng) – gây ra do các điện thế hoạt động ngắn hơn bình thường ở một số khu vực thất, hơn là toàn bộ tâm thất – làm nền tảng cho nhiều trường hợp VF nguyên phát [31]. Một điều thú vị, một số nghiên cứu về “hội chứng tái cực sớm” cũng báo cáo những bệnh nhân VF nguyên phát với sóng J cũng có khoảng QT ngắn hơn so với các nhóm đối chứng theo tuổi và giới tính [19, 32]. Mặt khác, thực tế chỉ có một số ít bệnh nhân mắc bệnh VF nguyên phát đã báo cáo bệnh sử gia đình có đột tử [3, 19, 33] là một lý luận mạnh mẽ chống lại vai trò các bệnh kênh ở tất cả các trường hợp.
Rối loạn nhịp thất trong VF nguyên phát thường được khởi kích (một cách bất biến)bằng các ngoại tâm thu thất có khoảng ghép rất ngắn [3 – 5], Haissaguerre đã cho thấy rõ các ngoại tâm thu có khoảng ghép ngắn này có nguồn gốc từ các sợi Purkinje [34, 35]. Các nhát bóp ngoại vi sợi Purkinje cũng được liên kết với khởi đầu VF ở bệnh tim thực thể[36], đặc biệt trong hoặc ngay sau khi nhồi máu cơ tim [37- 39]. Người ta có thể đề xuất các cơ chế rối loạn nhịp sau đây có thể giải thích các ngoại tâm thu có khoảng ghép ngắn có nguồn gốc từ các sợi Purkinje và khởi kích các VF nguyên phát: (1) vòng vào lại micro ở các chỗ nối cơ và Purkinje [40]; (2) hoạt động khởi kích được tạo ra khử cực sớm phase 3 trễ [41] do canxi không được thúc đẩy phát kích ở các sợi Purkinje và (3) phó tâm thu (là các ổ phát nhịp song song với nhịp xoang bình thường của tim, nhưng bình thường không bộc lộ ra có hiệu quả) có nguồn gốc từ các sợi Purkinje [42] phát ra một cách ngẫu nhiên vào sóng T của nhịp bình thường. Bất kể cơ chế nền nào phát ra các ngoại tâm thu Purkinje, nó đều có thời kỳ trơ của các cơ tim xung quanh ngắn, hoặc ở mức toàn bộ thất ở các bệnh nhân SQTS hoặc ở mức khu vực ở bệnh nhân có hội chứng tái cực sớm, cho phép các ngoại tâm thu được ghép sát để lan truyền và khởi kích VF.
Các biểu hiện lâm sàng
Các bệnh nhân có VF nguyên phát biểu hiện bằng hoặc ngất hoặc ngừng tim ở tuổi trưởng thành. Tuổi trung bình khi biểu hiện ở một số nhóm từ 35 – 45 tuổi và phần lớn trên 20 tuổi và trẻ hơn 65 tuổi khi biểu hiện [3, 19]. Hai phần ba (2/3) các bệnh nhân là nam giới [3, 19] .
Ngất do loạn nhịp thúc đẩy (Hình 1) và loạn nhịpkhi rung thất gây ngừng tim cũng tương tự về phương thức khởi phát, tần số thất và hình thái đa hình. Không rõ tại sao một số biến cố của VT đa hình ngừng một cách tự phát (gây ngất) trong khi những người khác lại thoái biến thành rung thất (gây ngừng tim) (hình 36.1b, c). Tuy nhiên, tỷ lệ bệnh nhân bị ngưng tim (khi đối lại với ngất) là cao hơn nhiều trong VF nguyên phát so với trong các bệnh kênh khác gây loạn nhịp thất đa hình như hội chứng QT dài hoặc CPVT [43]. Nói cách khác, rối loạn nhịp tim trong VF nguyên phát hiếm xẩy ra, nhưng một khi xảy ra, chúng thường dai dẳng và thường gây tử vong [28]. Theo nguyên tắc, ngất và ngưng tim trong VF nguyên phát không liên quan đến gắng sức hoặc căng thẳng cảm xúc [3, 19, 33]. Các rối loạn nhịp liên quan đến ngủ, phổ biến ở bệnh kênh ion natri (hội chứng Brugada và hội chứng QT dài type LQT3) hiếm khi xảy ra trong VF nguyên phát [3, 19, 33]. Cuối cùng, khoảng 25% bệnh nhân VF nguyên phát có các cơn bão loạn nhịp, tức là với các loạt VF (≥ 3 cơn) xảy ra trong vòng 24-48 giờ [3, 33, 44]. Một số loạt VF đã được sốt khởi kích [45].
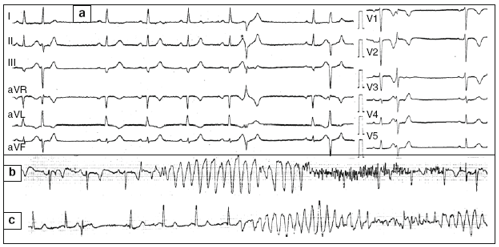
Hình 1. Ví dụ điển hình của VF nguyên phát. Người phụ nữ 54 tuổi này đã được giới thiệu để được tư vấn thần kinh vì “cơn động kinh tái phát”. Điện tâm đồ cơ bản của bà cho thấy nhịp xoang với các khoảng PR, QRS và QT bình thường (bản a). Tuy nhiên, có một số ngoại tâm thu thất với các khoảng ghép thay đổi, gồm các ngoại tâm thu khoảng ghép ngắn rơi vào đỉnh sóng T đi trước (*). Do các ngoại tâm thu khoảng ghép ngắn bệnh nhân được nhập khoa Tim Mạch (thay vì Khoa Thần kinh). Không bao lâu sau đó, cơn nhịp nhanh thất đa hình tự chấm dứt đã được ghi lại trong một trong những “cơn động kinh” (bản b). Rung thất đòi hỏi khử rung cũng được ghi lại ngay sau đó (bản c). Bệnh nhân được chẩn đoán là “rung tâm thất nguyên phát” và không có rối loạn nhịp thất trong khi điều trị với quinidine trong hơn 9 năm.
Điện tâm đồ
Điện tâm đồ (ECG) trong quá trình nhịp xoang thường được xác định như bình thường. Tuy nhiên, cần lưu ý khi có “VF nguyên phát” được xác định đầu tiên, ECG được coi là “bình thường” khi các bệnh lý được biết vào thời điểm đó đã được loại trừ [3]. Cụ thể, QT được coi là bình thường vì nó không kéo dài [3]. Tuy nhiên, như đã thảo luận ở trên, một tỷ lệ đáng kể các bệnh nhân có VF nguyên phát có khoảng QT “tương đối ngắn” vào thời điểm bắt đầu [18], các khoảng QT xuất hiện bình thường nhưng ngắn hơn so với những người đối chứng khỏe mạnh [19, 32] hoặc QT bình thường ở mức cơ bản nhưng không kéo dài đủ trong suốt thời gian nhịp tim chậm [26, 27]. Đỉnh sóng T đến khoảng cuối sóng T, là dấu hiệu của sự phân tán
tái cực và nguy cơ loạn nhịp trong hội chứng QT dài [46] và hội chứng Brugada [47] , là bình thường trong VF nguyên phát [18].
Mẫu tái cực sớm cũng được quan sát thấy ở những bệnh nhân VF nguyên phát thường xuyên hơn so với các đối chứng khỏe mạnh tương đương [19, 21, 48, 49] và một số sử dụng thuật ngữ “hội chứng tái cực sớm” để mô tả bệnh nhân có VF nguyên phát có mẫu ECG này [19, 49]. Trong hình thức đặc trưng nhất, tái cực sớm ở những bệnh nhân VF nguyên phát có hình thái sóng J tách biệt, sau đó là một phân đoạn ST đi ngang [50]. Dựa trên hàng loạt trường hợp đối chứng [21] và sử dụng các công thức xác suất điều kiện [21], người ta ước tính (1) tỷ lệ ước tính cho việc phát triển VF nguyên phát cho một cá nhân ở độ tuổi 35-45 là 3.4 trong 100.000; (2) nguy cơ tăng lên đến 11 trong 100.000 lần khi sóng J được xác định ở ECG và (3) nguy cơ tăng lên 30 trong 100.000 nếu sóng J được tiếp theo bằng đoạn ST đi ngang [50].
Các ngoại tâm thu thất hiếm khi xảy ra ở những bệnh nhân VF nguyên phát, nhưng khi xảy ra, chúng có khoảng ghép thay đổi với một số ngoại tâm thu ghép sát với phức bộ đi trước (khoảng ghép trung bình = 302 ± 52 ms trong hàng loạt trường hợp đã được quan sát [5], 297 ± 41 ms trong loạt trường hợp của Haissaguerre [35], 300 ± 35 ms trong loạt trường hợp của Champagne [51] và 340 ms trong loạt trường hợp của Nam [49]). Do khoảng cách ngắn, các ngoại tâm thu rơi vào đỉnh hoặc phần xuống của sóng T (Hình 36.1 và 36.2). Trong loạt trường hợp [5], tất cả các cơn VF bắt đầu bằng các ngoại tâm thất rơi vào phạm vi 40 ms (phần lớn trong phạm vi 20 ms) của đỉnh sóng T. Có vẻ như có một mối quan hệ nghịch giữa khoảng ghép của ngoại tâm thu và nguy cơ loạn nhịp ác tính với sự bùng nổ của VT đa hình dài hơn được khởi kích với các khoảng ghép ngăn hơn [35]. Tác giả Nam đã báo cáo loạn nhịp trong VF nguyên phát với tái cực sớm xảy ra trước các chu kỳ dài ngắn thường hơn trọng hội chứng Brugada [49]. Tuy nhiên, theo kinh nghiệm, rối loạn nhịp tim trong VF nguyên phát không phụ thuộc vào khoảng ngừng [3, 19, 33, 44, 51].
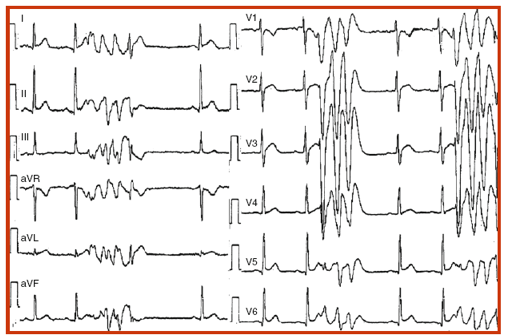
Hình 2. Kiểu điển hình của VF nguyên phát. Lưu ý khoảng ghép rất ngắn của các ngoại tâm thu khởi đầu nhịp nhanh thất đa hình. Cũng như, lưu ý mặc dù hình thái đa hình của rối loạn nhịp, khi nhiều hơn một cơn VT được ghi lại (như trong các chuyển đạo trước tim), các phức hợp đầu tiên, thứ hai và thứ ba của nhịp nhanh tương tự như nhau một cách rõ ràng.
Hình thái các ngoại tâm thu
Trong các thông báo đầu tiên cho thấy khởi phát của VF nguyên phát [3, 33, 52] , mẫu ECG của các ngoại tâm thu khoảng ghép ngắn đầu tiên tương tự nhau đã được quan sát, cụ thể là mẫu block nhánh bó trái và trục trái (hình 2). Tuy nhiên, các thông báo sau này [34, 35], cho thấy các ngoại tâm thu có mẫu khác, gợi ý các vị trí nguồn gốc có thể thay đổi. Điều thú vị, khi các cơn VT đa hình phức tạp được ghi 12 chuyển đạo, hình dạng của các nhát bóp khởi đầu của tất cả các cơn này là giống nhau. Điều này không chỉ áp dụng đối với phức hợp đầu tiên, mà còn cũng như với các phức hợp thứ hai và ba của các loại nhịp đa hình (hình 2). Quan sát gần đây ủng hộ ghi nhận VF nguyên phát có nguồn gốc ổ (xem dưới) [34, 35] .
Tư liệu điện sinh lý học
Các bệnh nhân VF nguyên phát có các khoảng A – H và H-V bình thường, cũng như các khoảng trơ thất của họ ở trong giới hạn bình thường [2, 53]. Điều này tương phản với các bệnh nhân hội chứng Brugada thường có khoảng H-V kéo dài [ 12 ] và các bệnh nhân hội chứng QT ngắn có các khoảng thời gian trơ ngắn ở nhĩ và thất [23, 54] .
Rối loạn nhịp thất được tạo ra bằng kích thất có chương trình là không thay đổi hình dạng đa hình, cụ thể VT đa hình hoặc VF (Fig. 36.3). VT đơn hình được tạo ra loại trừ chẩn đoán VT nguyên phát. Điều này trái người với các bệnh nhân hội chứng Brugada cũng có VF nguyên phát [55], nhưng hiếm có VT đơn hình [56–60] .
Tần số có khả năng tạo ra là chức năng giao thức (protocol) được sử dụng trong quá trình kích thích thất có chương trình. Nhiều nhà điện sinh lý không muốn rút ngắn khoảng ghép của các kích thích thất phóng ra vượt quá giá trị “danh nghĩa” là 200 ms. Điều này do nguy cơ “ngẫu nhiên” gây VF ở những người khỏe mạnh cũng tăng lên khi khoảng ghép của các kích thích thứ hai, thứ ba rút ngắn xuống dưới 200 ms [61-63]. Trên thực tế, trong các nghiên cứu nhỏ thực hiện cách đây 20 năm, 9% của các cá nhân khỏe mạnh – không có tư liệu hoặc nghi ngờ loạn nhịp thất nguyên phát – có VF có thể tạo ra khi khoảng ghép được giới hạn bằng các khoảng thời gian trơ của thất [61- 63]. Hơn nữa, mới đây thêm 40% các nhóm đối chứng người khỏe mạnh có VT đa hình tạm thời có thể tạo ra và điều này đưa đến các giao thức kích thích nhịp sớm không liên tục [61- 63]. Do đó, người ta cần ghi nhận ít nhất 9% người khỏe mạnh sẽ có thể tạo VF nếu giao thức kích ngoài tiến triển (với kích thích ngoài hai (kép) và ba có khoảng ghép ngắn hơn 200 ms) được sử dụng [64]. Mặt khác, theo kinh nghiệm của các nhà nghiên cứu, có đến 80% bệnh nhân với VF nguyên phát có VF có thể tạo ra với các giao thức kích thích ngoài tiến triển gồm kích thích thất ngoài kép và ba tại hai vị trí tâm thất phải và sử dụng lặp lại của kích thích ngoài ở khoảng ghép ngắn nhất bắt được tâm thất [2, 52, 53]. Tần số có thể tạo ra rất cao này gợi ý khả năng tạo ra VF, với giao thức kích thích ngoài tiến triển, là tiêu chí có giá trị của kích thích thất có chương trình, do đó có thể được sử dụng cho hướng dẫn điều trị rối loạn nhịp bằng quinidine ở bệnh nhân VF nguyên phát…
Gần đây, Haissaguerre và cộng sự thực hiện ghi nội mạc tim ở bệnh nhân VF nguyên phát vào thời điểm khi họ có các ngoại tâm thu thất tự phát thường xuyên và/hoặc bùng phát VT đa hình [34, 35]. Các nhà nghiên cứu đã xác định vị trí nguồn gốc của các loạn nhịp thất này ở 27 bệnh nhân. Định khu thành công vị trí nguồn gốc của rối loạn nhịp thất được hướng dẫn bằng ghi các đường ghi nội tâm mạc rất sớm và đã khẳng định bằng cách loại bỏ (abolition) các rối loạn nhịp thất tiếp theo triệt phá ổ khởi phát bằng năng lượng tần số radio. Điện thế của Purkinje được ghi ở vị trí xuất phát của rối loạn nhịp thất ở 23/27 bệnh nhân này (85%) (ở vách liên thất trái 10 bệnh nhân, thất phải trước 9 bệnh nhân và ở cả hai vị trí 4 bệnh nhân). Các điện thế Purkinje đi trước hoạt động của cơ tim khu vực bằng khoảng 11 ± 5 giây trong quá trình nhịp xoang và 10 ± 150 ms trong quá trình ngoại vị thất nguyên phát [34, 35]. Trên cơ sở ghi nội tâm mạc này, dường như rối loạn nhịp trong VF nguyên phát có nguồn gốc khu trú và các ổ khởi phát trong phạm vi các sợi Purkinje ở đa số bệnh nhân. Lưu ý, ổ phát kích thích không nằm trong mạng Purkinje chỉ ở 4 bệnh nhân (15%) và ở tất cả những bệnh nhân này, rối loạn nhịp tim có nguồn gốc từ đường ra thất phải (RVOT). Có thể là VF nguyên phát có nguồn gốc từ RVOT và “biến thể ghép ngăn của RVOT” do một nhóm nghiên cứu mô tả [65], và “VT RVOT nguyên phát ác tính” (malignant idiopathic RVOT-VT) do Noda [66] mô tả hoàn toàn giống nhau.
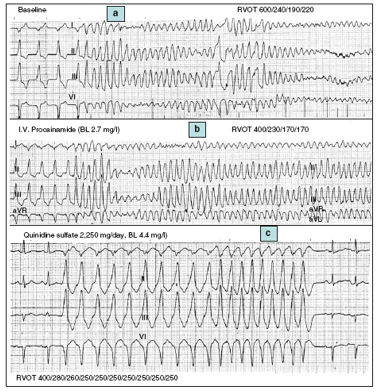
Hình 3. Các kết quả điển hình nghiên cứu điện sinh lý trong VF nguyên phát. Bảna: Trong nghiên cứu cơ bản, VF được gây ra bằng ba kích thích thất ngoài với các khoảng ghép ngắn từ đường ra thất phải. Tạo nhịp thất cơ bản ở 100 nhịp / phút (chu kỳ 600 ms) được theo sau bằng ba phần kích thích ngoài 240, 190 và 220 ms và điều này khởi đầu VF đòi hỏi phải sốc bằng dòng điện một chiều để cắt VF. Bản b: Sau khi tiêm tĩnh mạch lên đến 1.000 mg procainamide, protocol kích thích ngoài được lặp lại và VF một lần nữa được tạo ra. Lưu ý mặc dù có các mức độ điều trị của procainamide, thời gian trơ có hiệu quả là ngắn đủ để cho phép bắt được thất với các khoảng ghép ngắn (230, 170 và 170 ms cho kích thích lần thứ nhất, thứ hai và thứ ba). Bản c. Sau 5 ngày điều trị với quinidin bằng đường uống, không còn có thể bắt được tâm thất với các khoảng ghép ngắn.
Chẩn đoán
Chẩn đoán VF nguyên phát ở người ngừng tim sống sót là đơn giản khi khởi phát đầu tiên của VT/VF đa hình tự phát được ghi nhận (thường trong quá trình bão loạn nhịp) và điều này cho thấy khởi đầu của VT/VF đa hình bằng các ngoại tâm thu thất khoảng ghép rất ngắn (hình 3) [ 3, 5 ]. Điều này chỉ do ba trạng thái khác nhau đưa đến kiểu đặc trưng của khởi đầu VF (thiếu máu cơ tim cục bộ [ 67, 68], [ 69 ] và hội chứng QT ngắn [ 117 ] ) có thể được nhận biết với các test phù hợp.
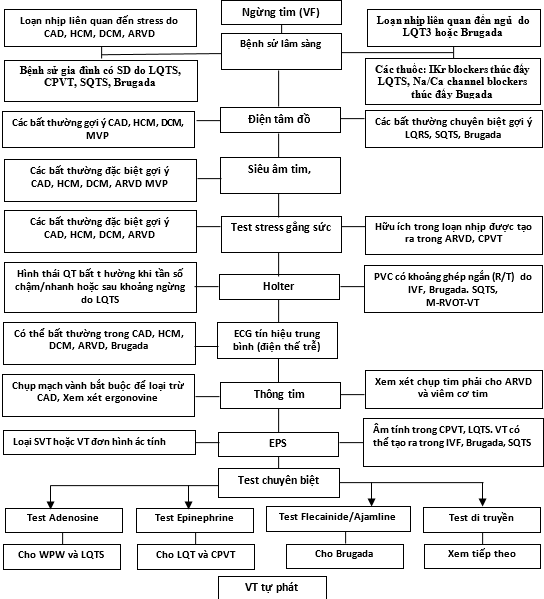
Hình 4. Thuật toán cơ bản được đề nghị cho chẩn đoán VF nguyên phát. CAD: bệnh mạch vành, HCM: bệnh cơ tim phì đại, DCM: bệnh cơ tim giãn. ARVD: Dị sản thất phải gây loạn nhịp. LQTS: hội chứng QT dài, CPVT: nhịp nhanh thất do cathecholamine. LQT3: Hội chứng QT dài type 3. SQTS: hội chứng QT ngắn. MVP: sa van hai lá. CT: chụp cắt lớp điện toán. MRI: hình ảnh cộng hường từ. M-RVOT-VT: hình thái ác tính của nhịp nhanh thất nguồn gốc đường ra thất phải. WPW: hội chứng Wolff-Parkinson-White..IVF: rung thất nguyên phát. VT: nhịp nhanh thất. SVT: nhịp nhanh trên thất.
Tuy nhiên, thường xuyên hơn, bệnh nhân được nhập viện sau khi hồi phục từ ngừng tim và có rung thất được chứng minh bằng tư liệu, nhưng không có khả năng ghi được sự khởi phát rối loạn nhịp. Trong những trường hợp như vậy, việc chẩn đoán VF nguyên phát được xác lập bằng cách loại trừ tất cả các nguyên nhân có thể nhận diện được và được ủng hộ tiếp theo bằng khả năng tạo ra VF với kỹ thuật kích thích thất co chương trình. Một cuộc thảo luận về tất cả các nguyên nhân tiềm tàng của đột tử sẽ trình bày ở một chuyên đề khác, nhưng cách tiếp cận trong thực hành được trình bày trong hình minh họa.
Chẩn đoán VF nguyên phát nên được xem xét ở những bệnh nhân có hiện tượng ngất mà không có loạn nhịp được chứng minh bằng tư liệu. Thực tế cho thấy, cần nhấn mạnh trong đa số bệnh nhân có hiện tượng ngất khi không có bệnh tim, chẩn đoán ngất do cường phế vị (chứ không phải ngất do loạn nhịp) sẽ rõ ràng là từ bệnh sử lâm sàng. Ngoài ra, đại đa số bệnh nhân có ngất dường như không có nguồn phế vị cũng có những bất thường về điện tâm đồ hoặc siêu âm tim sẽ gợi ý một chẩn đoán cơ bản. Do đó, chỉ có rất ít được kiểm chứng bằng bệnh nhân biểu hiện ngất ở họ có biểu hiện bệnh sử đầy đủ gợi ý nguồn gốc rối loạn nhịp, tuy nhiên tất cả các nghiên cứu không xâm lấn, gồm gắng sức [70-72] và các test tư thế đứng [73] để loại trừ rối loạn nhịp thúc đẩy do gắng sức và hội chứng QT dài, [74, 75] cùng với thử thách bằng thuốc để loại trừ hội chứng Brugada [ 74, 75 ] hội chứng QT dài [76, 77 ] và Wolff Parkinson White [78] đều âm tính. Trong những trường hợp như vậy, đánh giá điện sinh lý có thể được thực hiện để loại trừ block nội bó His như là nguyên nhân gây ngất. Tuy nhiên, khuyến cáo việc thực hiện kích thích thất có chương trình đối với bệnh nhân ngất không có bằng chứng bệnh tim và không có rối loạn nhịp được chứng minh bằng tư liệu (đặc biệt ngoại tâm thu thất khoảng ghép rất ngắn là một vấn đề. Điều này do, nếu không có bệnh tim, loạn nhịp thất (nếu có) có thể có hình thái đa hình. Hiểu được ý nghĩa của VF khi không có các rối loạn nhịp nguyên phát được chứng minh là khó xảy ra, vì các rối loạn nhịp như vậy có thể gây ra ở ít nhất 9% người khỏe mạnh [64]. Do đó, việc thực hiện kích thích thất có chương trình chỉ nên được thực hiện khi cả bác sĩ và bệnh nhân chuẩn bị để chấp nhận sự khởi phát của VF như một đáp ứng “dương tính”.
Chẩn đoán phân biệt
Các hình thái tinh tế của Bệnh Tim Thực thể(subtle forms of organic heart disease)
Loại trừ tất cả các dạng bệnh tim thực thể là cần thiết trước khi chẩn đoán VF nguyên phát. Tuy nhiên, cần lưu ý một số dạng của bệnh tim có thể gây ra rối loạn nhịp thất ác tính ở một thời điểm khi các bất thường giải phẫu là tối thiểu và khó phát hiện bằng các phương pháp hình ảnh. Ví dụ, bệnh nhân bệnh cơ tim phì đại do đột biến Troponin T có thể có nguy cơ tử vong do rối loạn nhịp ở thời điểm khi phì đại thất trái còn chưa rõ [79, 80]. Tương tự, loạn sản xuất thất phải đôi khi được xác định là nguyên nhân gây tử vong đột ngột chỉ trong suốt cuộc kiểm tra pháp y và mặc dù đã thực hiện rộng rãi các chẩn đoán âm tính [81]. Lưu ý, những bất thường về giải phẫu học tinh tế, như sa van hai lá không có ý nghĩa huyết động, không nhất thiết phải được chấp nhận như là nguyên nhân gây ra ngừng tim. Mặt khác, các dấu hiệu rối loạn chức năng tâm thất trái sau khi hồi sức không nhất thiết phải được sử dụng để loại trừ chẩn đoán VF nguyên phát vì hồi sức kéo dài có thể dẫn đến những bất thường về điện tâm đồ và siêu âm tim không thể phân biệt được với bệnh nhân bệnh cơ tim giãn [82, 83]. Nếu những bất thường như vậy được giải quyết, cần phải xem xét chẩn đoán VF nguyên phát.
Hội chứng Wolff-Parkinson-White
Bệnh nhân có đường phụ nhĩ thất có thể có kích thích sớm tối thiểu hoặc không (có thể có các phức hợp QRS hẹp) nếu chúng cũng có dẫn truyền nhanh qua nút AV hoặc nếu các đường phụ khu trú ở thành bên trái (cách xa nút xoang). Tuy nhiên, các đường này có thể có thời gian trơ ngắn. Các bệnh nhân như vậy có thể phát triển rung nhĩ với tần số thất nhanh và có thể thoái hóa thành rung thất (VF). Nếu rung nhĩ được kích thích sớm không được ghi nhận và bệnh nhân được nhận thấy trong VF, điện tâm đồ gần bình thường trong quá trình nhịp xoang có thể đưa đến chẩn đoán nhầm “rung thất nguyên phát” do tất cả các test hình ảnh học sẽ là bình thường. Chẩn đoán sai về VF nguyên phát có thể được tăng thêm tiếp theo sự hỗ trợ từ nghiên cứu điện sinh lý nếu kích thích nhĩ không được thực hiện trước khi tạo nhịp thất do kích thích thất có chương trình nhiều khả năng để tạo ra VF ở các bệnh nhân WPW [84]. Do đó, loại trừ các đường phụ – hoặc bằng tiêm adenosine như test cạnh giường hoặc bằng tạo nhịp nhĩ trong quá trình nghiên cứu điện sinh lý – là bước bắt buộc trong quá trình khám các bệnh nhân VF sống sót, ngay cả khi điện tâm đồ được đánh giá là “bình thường”. Lưu ý, các trường hợp ngừng tim hiếm có gây ra do nhịp nhanh trên thất rất nhanh ở các bệnh nhân không có hội chứng Wolff-Parkinson-White cũng đã được mô tả [85].
Nhịp nhanh thất đa hình nhạy cảm với catecholamine
Các bác sĩ có thể bỏ qua việc thực hiện test gắng sức ở các người sống sót sau ngừng tim do chụp mạch vành sẽ bộc lộ tổn thương mạch vành có ý nghĩa. Người ta đã gặp các bệnh nhân bị CPVT được chẩn đoán sai là “VF nguyên phát” chỉ vì không thực hiện test gắng sức. Từ tất cả test khác, gồm các nghiên cứu về điện sinh lý, đều bình thường trong bệnh này, test stress gắng sức là test bắt buộc ở các bệnh nhân sống sót sau ngừng tim. Mặc dù phần lớn bệnh nhân có CPVT có đáp ứng bệnh lý với gắng sức (rung nhĩ được thúc đẩy do gắng sức tiếp theo các ngoại tâm thu thất đa ổ của chúng, VT hai chiều và VT đa hình), người ta mới ghi nhận gần đây một số bệnh nhân có CPVT đã được chứng minh về mặt di truyền chỉ có các ngoại tâm thu thất đơn nhìn giống như “các ngoại tâm thu đường ra thất phải lành tính” (RVOT) trong quá trình gắng sức tối đa [86, 87]. Vừa mới đây, một gia đình có CPVT trong đó các cá thể bị ảnh hưởng có test stress gắng sức âm tính nhưng có thể nhận biết bằng sự kéo dài của QT sau khoảng ngừng đáng kể, đã được mô tả [88]. Các bệnh nhân như vậy có thể dễ dàng chẩn đoán nhầm như “VF nguyên phát”.
Hội chứng QT dài (LQTS)
Các khoảng QTc của dân số khỏe mạnh, cũng như QTc của bệnh nhân với LQTS có phân bố bình thường và có sự chồng chéo đáng kể giữa QTc của cả hai quần thể. Quan trọng hơn, 12% bệnh nhân có LQTS đã được chứng minh về mặt di truyền có “QT bình thường” khi được xác định là QTc < 440ms [89]. Việc xác định bệnh nhân có LQTS có QT giới hạn là một thách thức đặc biệt trong kiểu gen LQT1 do hình thái sóng T, chúng thường bất thường trong LQT2 và LQT3, đại đa số bình thường trong LQT1. Thật may, test thử nghiệm epinephrine đặc biệt có hiệu quả đối với các đáp ứng bất thường bộc lộ ra ở LQT1 [76, 77]. Gần đây người ta đã mô tả cơn nhịp nhanh đột ngột được gây ra bằng kéo dài LQTS bị che lấp ở các trường hợp giới hạn và sử dụng như test đầu giường [73].
Hội chứng QT ngắn (SQTS)
SQTS mới được mô tả [15, 16, 23] được gây ra do các đột biến gene liên quan đến các kênh kali tương tự gây ra LQTS nhưng với hiệu quả ngược lại [90-93]. Nói cách khác, trong SQTS có quá nhiều dòng kali đi ra ngoài, làm rút ngắn khoảng thời gian điện thế hoạt động và thời gian trơ có hiệu quả của thất. Phân biệt VF nguyên phát với SQTS không dễ dàng. Mặc dù các trường hợp đầu tiên của SQTS có các khoảng QT cực ngắn (QTc ngắn hơn 300 ms)[15, 16, 23 ], gần đây các trường hợp được mô tả của SQTS đã được chứng minh về gene có các khoảng QTc 360 ms [25]. Thật vậy, người ta mới chỉ ra các khoảng QT “ngắn tương đối” (QTc <360 ms) thường được quan sát ở những đàn ông khỏe nhưng thường hơn một cách tính toán ở các đàn ông bị VF nguyên phát [18]. Hơn nữa, những bệnh nhân VF nguyên phát có các khoảng QT bình thường ở nhịp tim bình thường, nhưng QT không được kéo dài khi tần số tim của họ chậm xuống, dẫn đến các giá trị QTc một cách bất thường trong quá trình nhịp chậm [26, 27]. Cuối cùng, bệnh nhân SQTS và bệnh nhân VF nguyên phát có các đặc điểm lâm sàng sau: (1) cả hai nhóm bệnh nhân đều nguyên phát [5,17] và rối loạn nhịp thất có thể tạo ra [3,23]; (2) cả hai nhóm bệnh nhân dường như đều đáp ứng đặc biệt tốt với điều trị quinidine (33, 52, 54, 94, 95); (3) cả hai nhóm bệnh nhân đều ở nguy cơ sốc ICD không phù hợp do quá nhạy cảm của sóng T trong buồng tim [96, 97]. Bệnh nhân có SQTS có thể bị chẩn đoán nhầm là “VF nguyên phát” nếu khoảng QT chỉ được đo khi nhịp tim tương đối nhanh. Điều này do vấn đề chính trong SQTS là sự thất bại của kéo dài QT phù hợp trong quá trình nhịp chậm [23].
Hội chứng Brugada
Chúng tôi ước tính cứ 5 bệnh nhân được chẩn đoán ban đầu “VF nguyên phát” có một bệnh nhân thực chất là hội chứng [13] và số liệu tương tự cũng được các tác giả khác báo cáo [98]. Hơn nữa, nếu tất cả các bệnh nhân VF nguyên phát được thực hiện test một cách có hệ thống bằng ECG [99] và ghi Holter ‘[100] với các điện cực trước ngực phải được đặt ở các vị trí cao hơn, cũng như các test thử thách thuốc sử dụng chẹn kênh natri, khoảng 40% VF nguyên phát có thể được chẩn đoán là hội chứng Brugada [101].
Sự thay đổi Khoảng Ghép Ngắn của Nhịp nhanh Đường ra Thất Phải
Đường ra thất phải (RVOT) là vị trí nguồn gốc của type phổ biến nhất của nhịp nhanh thất (VT) xuất hiện ở các bệnh nhân không có bệnh tim thực thể [33]. RVOT-VT hình thái khác biệt này (các phức bộ QRS với mẫu block nhánh trái và các sóng R cao ở các chuyển đạo dưới) và nói chung, ít đưa đến tổn thương huyết động. Chúng tôi đã gặp một bệnh nhân được chẩn đoán ngất theo dõi do động kích đã điều trị và theo dõi hơn 3 năm tại Viện Tim thành phố HCM, nhưng thực chất là RVOT-VT, nhờ bắt được cơn ngất đang ghi ECG Holter, cuối cùng đã được triệt phá qua catheter bằng năng lượng tần số radio thành công, theo dõi hơn 10 nay không tái phát. Do đó, RVOT-VT được coi là rối loạn nhịp lành tính [33]. Tuy nhiên, các nhóm của Noda và Shimizu [65] và nhóm Noda và Shimizu [65, 66] mới mô tả các bệnh nhân gần đây với “ngoại vị RVOT lành tính” điển hình về phương diện khác đã tiếp tục phát triển VF nguyên phát hoặc VT đa hình. Người ta còn chưa rõ nếu các bệnh nhân có VF nguyên phát và các bệnh nhân với hình thái được mô tả mới này của “VT đa hình” từ RVOT mô tả các khía cạnh khác nhau của một bệnh hay hai rối loạn khác biệt [102]. Tuy nhiên, một số đặc điểm khác nhau giữa hai nhóm: (1) RVT-VT đơn hình điển hình khác cũng được thấy ở những bệnh nhân RVOT-VT đa hình ác tính [65,66] nhưng không bao giờ được thấy trong VF nguyên phát. (2) Chỉ có 5% bệnh nhân với “VT đa hình ác tính” có VF có thể tạo ra [65, 66] bằng kích thích thất có chương trình, trong khi đa số bệnh nhân VF nguyên phát có VF có thể tạo ra [33, 52]. (3) Khoảng ghép của các ngoại tâm thu thất khởi đầu rối loạn nhịp thất ác tính cách liên kết của các trường hợp ngoại tâm thu thất bắt đầu các rối loạn nhịp thất ác tính là rất ngắn không thể thay đổi trong VF nguyên phát [3, 5] nhưng lại dài hơn, thay đổi từ “tương đối ngắn” [65] đến “bình thường” [66] trong “RVOT-VT đa hình” [103]. Quan sát cuối cùng phù hợp với kết quả lập bản đồ trong buồng tim của Haissaguerre [35]: Trong loạt nghiên cứu này, VF nguyên phát bắt nguồn từ Purkinje ở 86% bệnh nhân và từ RVOT trong 14% còn lại. Một lần nữa, khoảng ghép của ngoại tâm thu khởi đầu VF dài hơn đối với rối loạn nhịp nguồn gốc trong RVOT so với rối loạn nhịp được khởi kích bằng các sợi Purkinje (355 ± 30 ms so với 280 ± 26 ms, p <0,01) [35].
Tiên lượng và Điều trị VF Nguyên phát
Tần số xuất hiện rối loạn nhịp thất ác tính trong VF nguyên phát, nếu không điều trị, là không cao. Trong theo dõi trung bình 6 năm, hơn 40% bệnh nhân có VF tái phát và nguy cơ cao hơn đối với những người có điện tâm đồ bình thường (nghĩa là sau khi loại trừ những người có thể hội chứng Brugada) [104]. Trong một loạt các bệnh nhân mới đây “VF nguyên phát thật sự” (có nghĩa là, loại trừ không chỉ những bệnh nhân ECG type Brugada ở cơ bản nhưng cũng bệnh nhân chênh lên đoạn ST được phát triển khi được thử thách với chẹn kệnh natri), nguy cơ VF tái phát là 39% trong 3,4 ± 2,3 năm [51]. Do đó, mỗi lần chẩn đoán VF nguyên phát được thực hiện, một số hình thức của điều trị là bắt buộc. Điều trị có thể gồm cấy ICD, liệu pháp dùng thuốc với quinidin, triệt phá bằng tần số radio các ổ khởi phát hoặc phối hợp các biện phát trên.
Điều trị thuốc với Quinidine
Các bệnh nhân đầu tiên bị VF nguyên phát được mô tả trong tài liệu, vào năm 1929 [1] và năm 1949 [105], đã được điều trị bằng quinidine sau khi nhiều đợt VT VT và VF đã được chứng minh bằng tư liệu. Cả hai bệnh nhân đều đáp ứng hoàn toàn [1, 105]. Trên thực tế, một ấn phẩm thứ hai báo cáo việc theo dõi lâu dài các bệnh nhân ban đầu được báo cáo vào năm 1949, cho thấy bệnh nhân này cuối cùng đã chết vì bệnh ung thư ở tuổi già, mà không gặp tái phát loạn nhịp khi dùng thuốc quinidine trong 40 năm [106]. Năm 1987, Belhassen đi tiên phong trong điều trị VF nguyên phát với quinidine được hướng dẫn bằng EPS sau khi quan sát thấy VF có thể dễ dàng có thể tạo ra ở trạng thái cơ bản như không thể tạo ra sau điều trị quinidine [2]. Lưu ý, một trong các bệnh nhân bao gồm trong báo cáo đầu tiển [52], đã hoàn thành trên 25 năm ổn định được điều trị theo hướng dẫn điện sinh lý bằng amiodarone và quinidine sau khi trải qua các cơn bão rối loạn nhịp VF khi không điều trị và ngất tái phát khi điều trị amiodarone đơn thuần [94]. Kinh nghiệm với điều trị quinidine được xuất bản năm 1999 [ 53 ].
Năm 1990, khi lần đầu tiên xem xét lại chủ đề VF nguyên phát [3], người ta phát hiện ra tỷ lệ ngừng tim tái phát tăng lên rất cao trong quá trình điều trị bằng các thuốc chống loạn nhịp khác (bao gồm amiodarone, thuốc chẹn bêta hoặc verapamil) [3]. Tỷ lệ tăng loạn loạn nhịp với verapamil rất đáng chú ý vì thuốc đó được Leenhardt và Coumel đề xuất để điều trị “biến thể xoẵn đỉnh khoảng ghép ngắn” [4], một thực thể có thể đại diện cho VF nguyên phát. Ngược lại, người ta thấy không có tỷ lệ tái phát với quinidine [3]. Vào thời điểm ICD trở nên sẵn có trên thị trường, người ta tiếp tục khuyên dùng quinidine như là liệu pháp duy nhất cho những bệnh nhân được chọn lọc một cách thích hợp với VF nguyên phát, bao gồm cả những bệnh nhân hồi phục sau khi ngừng tim nguyên phát [107]. Tiêu chuẩn người ta đưa ra đối với liệu pháp quinidine ở những người sống sót sau VF gồm tất cả những điều sau: (1) Chẩn đoán VF nguyên phát có hoặc không có hội chứng Brugada; (2) VF có thể được tạo ra khi không có thuốc với kích thích thất có chương trình (Hình 36.3); (3) rối loạn nhịp tim không thể gây ra trong quá trình điều trị uống quinidine mặc dù có một quy trình kích thích tâm thất rất mạnh (hình 36.3) [2, 108, 109] (4) sự đồng ý của bệnh nhân được thông báo rõ về nguy cơ và lợi ích của ICD và liệu pháp quinidine cho bệnh này [107]; (5) khẳng định lặp đi lặp lại sự tuân thủ của thuốc trong quá trình theo dõi lâu dài (sự tuân thủ được đánh giá bằng mức độ quinidin huyết thanh và ảnh hưởng quinidine trên khoảng QT). Kết quả của sử dụng cách tiếp cận này đã được xuất bản năm 1999 [53]. Các kết quả này được thể hiện trong hình 36.5 và có thể được tóm tắt như sau: Trong số 34 bệnh nhân bị VF nguyên phát (tất cả sau hồi sức ngừng tim), 26 (80%) có VF có thể tạo ra trong nghiên cứu điện sinh lý cơ bản và tất cả trừ một không thể kích thích tạo ra VF với quinidine. Tác dụng phụ của quinidin dẫn đến ngưng dùng quinidine ở 14% bệnh nhân. Tuy nhiên, 23 bệnh nhân (2 trong số 3 bệnh nhân từ nhóm nghiên cứu ban đầu của những người sống sót sau khi bị ngừng tim) vẫn tiếp tục điều trị bằng quinidine (không có ICD dự phòng) và tất cả đều sống và hoàn toàn không có các triệu chứng rối loạn nhịp, đến bây giờ vượt quá 10 năm. Hiệu quả lâu dài của quinidine để ngăn ngừa có thể tạo ra VF đã được khẳng định [110]. Tương đối sớm theo kinh nghiệm của các tác giả, ba bệnh nhân đã nghiên cứu điện sinh lý âm tính không có thuốc đã nhận được quinidine theo kinh nghiệm. Tất cả những bệnh nhân này đã chết 4-8 năm sau cơn VF đầu tiên, 3 bệnh nhân này ngưng theo dõi lâu trước khi họ chết. Do đó, người ta không biết liệu những người tử vong do sự tuân thủ kém hoặc do sự thất bại thất thường như vậy liên quan đến thực tế điều trị quinidine cho các bệnh nhân đặc biệt này là theo kinh nghiệm và không được hướng dẫn bằng nghiên cứu điện sinh lý (vì 3 bệnh nhân này không thể tạo ra VF về cơ bản). Tuy nhiên, người ta không còn khuyến cáo sử dụng quinidine theo kinh nghiệm cho những bệnh nhân không thể tạo ra VF sau ngừng tim nguyên phát, phân nhóm các bệnh nhân đối với họ cấy ICD là bắt buộc. Tuy nhiên, người ta đã sử dụng quinidine thành công để kiểm soát các cơn bão rối loạn nhịp tim ở những bệnh nhân ban đầu đã nhận được ICD hoặc do không có khả năng tạo ra VF trong nghiên cứu điện sinh lý cơ bản hoặc do (rất hiếm) sự dai dẳng của khả năng tạo ra trong khi điều trị quinidine. Đáp ứng tuyệt vời của cơn bão VF trong VF nguyên phát với hội chứng Brugada cũng đã được thông báo nhắc lại [60, 111-111]. Lưu ý, những người tử vong xảy ra 4-8 năm sau khi sự kiện VF đầu tiên chứng minh rõ ràng những bệnh nhân VF nguyên phát vẫn có nguy cơ bị loạn nhịp gây tử vong-tái phát ngay cả sau những giai đoạn không triệu chứng kéo dài. Do đó, các giai đoạn không triệu chứng dài sau khi cơn VF đầu tiên không được giải thích như sự thoái lui của “viêm cơ tim” không được nhận biết và không thể được coi là “dấu hiệu tiên lượng tốt”.
Triệt phá bằng tần số radio
Triệt phá các ổ khởi kích bằng tần số radio trên cơ sở catheter ngày nay là một phương thức điều trị được chấp nhận cho rung nhĩ. Haissaguerre [35, 44] và các tác giả khác [66, 114, 115] đã sử dụng khái niệm tương tự để điều trị VF nguyên phát. Hình thức trị liệu này đã được sử dụng chủ yếu để điều trị bệnh nhân đã được cấy ICDs những nhận được nhiều cú sốc ICD do các cơn bão loạn nhịp. Sự triệt phá thành công đầu tiên đã được Aizawa báo cáo vào năm 1992 [114] trong khi các báo cáo của Haissaguerre [35] và Noda và Shimizu [66] đã báo cáo một loạt tương đối lớn. Các loạt của Haissaguerre [35] và Noda [66] khác nhau về vị trí xuất phát của đích rối loạn nhịp: Noda nhắm mục tiêu VT đa hình có nguồn gốc từ đường ra thất phải [66]. Ngược lại, 85% số rối loạn nhịp thất đa hình do Haissaguerre triệt phá đã được vẽ bản đồ ở hệ thống Purkinje ở thất trái hoặc phải, trong khi vị trí xuất phát của VF nằm ở đường ra thất phải chỉ có 4 bệnh nhân (15%) [35]. Triệt phá thành công cấp thời đã đạt được ở tất cả các trường hợp trong khi 24 bệnh nhân (89%) không tái phát VF đã không dùng thuốc trong thời gian theo dõi. Tuy nhiên, nguy cơ tái phát VF nguyên phát trong quá trình theo dõi dài hạn là 18% [116], chứng tỏ việc triệt phá bằng tần số radio không nên được coi là “chữa bệnh” hoặc thay thế cấy ICD.
Cấy ICD
Không còn nghi ngờ ICD cung cấp điều trị hiệu quả nhất để ngăn ngừa tử vong do VF tự. Thật vậy, cấy ICD được coi là “liệu pháp duy nhất có hiệu quả” cho VF nguyên phát được hầu hết các tác giả nêu ra. Tuy nhiên, khi so sánh cấy ICD với điều trị quinidine cho VF nguyên phát, cũng nên xem xét các tác dụng phụ tiềm tàng của tất cả các can thiệp này.
Trong AVID, một nghiên cứu đa trung tâm lớn về cấy ICD cho loạn nhịp thất ác tính ở bệnh nhân có bệnh tim thực thể [117], nguy cơ các biến cố bất lợi, đủ nghiêm trọng để đảm bảo sự can thiệp lại, là 12% [118]. Vì chỉ có các chuyên gia điện sinh học có kinh nghiệm từ các trung tâm có uy tín tham gia AVID, tỷ lệ biến chứng 12% này có thể là một ước tính vừa phải. Hơn nữa, tỷ lệ biến chứng do cấy ICD trong VF nguyên phát có thể cao hơn 12% được báo cáo trong AVID. Điều này là do các bệnh nhân trong AVID tương đối lớn tuổi (65 tuổi ± 11 tuổi) [117] và có tần số tử vong 25% trong 3 năm mặc dù ICD có liên quan đến bệnh tim nền thực thể của họ [117]. Ngược lại, những bệnh nhân VF nguyên phát trẻ hơn một cách có ý nghĩa và có nguy cơ vô cùng thấp cho tử vong không phải do rối loạn nhịp tim. 28% nguy cơ biến chứng dài hạn sau khi cấy ICD cho hội chứng Brugada [119] là đại diện hơn của nguy cơ cho các bệnh nhân VF nguyên phát.
Tài liệu tham khảo
1. Dock W. Transitory ventricular fi brillation as a cause of syncope and its prevention by quinidine sulfate. Am Heart J. 1929;4:709–14.
2. Belhassen B, Shapira I, Shoshani D, Paredes A, Miller H, Laniado S. Idiopathic ventricular fi brillation: inducibility and bene fi cial effects of class I antiarrhythmic agents. Circulation. 1987;75: 809–16.
3. Viskin S, Belhassen B. Idiopathic ventricular fi brillation. Am Heart J. 1990;120:661–71.
4. Leenhardt A, Glaser E, Burguera M, Nurnberg M, Maison-Blanche P, Coumel P. Short-coupled variant of torsade de pointes. A new electrocardiographic entity in the spectrum of idiopathic ventricular tachyarrhythmias. Circulation. 1994;89:206–15.
5. Viskin S, Lesh M, Eldar M, et al. Mode of onset of malignant ventricular arrhythmias in idiopathic ventricular fi brillation. J Cardiovasc Electrophysiol. 1997;8:1115–20.
6. Haissaguerre M, Extramiana F, Hocini M, et al. Mapping and ablation of ventricular fi brillation associated with long-QT and Brugada syndromes. Circulation. 2003;108:925–8.
7. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59–68.
8. Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare delleta pediatrica. Clin Pediatr. 1963;45:656–83.
9. Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–6.
10. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc D, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year followup of 21 patients. Circulation. 1995;91:1512–19.
11. Aponte G. The enigma of “Bangungut”. Ann Intern Med. 1960;52:1258–63.
12. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20:1391–6.
13. Viskin S, Fish R, Eldar M, et al. Prevalence of the Brugada sign in idiopathic ventricular fi brillation and healthy controls. Heart. 2000;84:31–6.
14. Nademanee K, Veerakul G, Nimmannit S, et al. Arrhythmogenic marker for the sudden unexpected death syndrome in Thai men. Circulation. 1997;96:2595–600.
15. Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99–102.
16. Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965–70.
17. Schimpf R, Bauersfeld U, Gaita F, Wolpert C. Short QT syndrome: successful prevention of sudden cardiac death in an adolescent by implantable cardioverter-de fi brillator treatment for primary prophylaxis. Heart Rhythm. 2005;2:416–17.
18. Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fi brillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fi brillation and healthy controls. Heart Rhythm. 2004;1:587–91.
19. Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358:2016–23.
20. Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med. 2008;358:2078–9.
21. Rosso R, Kogan E, Belhassen B, et al. J-point elevation in survivors of primary ventricular fibrillation and matched control subjects incidence and clinical signi fi cance. J Am Coll Cardiol. 2008;52:1231–8.
22. Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7:549–58.
23. Gussak I, Bjerregaard P. Short QT syndrome–5 years of progress. J Electrocardiol. 2005;38:375–7.
24. Viskin S. The QT, interval: too long, too short or just right. Heart Rhythm. 2009;6:711–15.
25. Templin C, Ghadri JR, Rougier JS, et al. Identi fi cation of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur Heart J. 2011;32:1077–88.
26. Fujiki A, Sugao M, Nishida K, et al. Repolarization abnormality in idiopathic ventricular fi brillation: assessment using 24-hour QT-RR and QaT-RR relationships. J Cardiovasc Electrophysiol. 2004; 15:59–63.
27. Sugao M, Fujiki A, Sakabe M, et al. New quantitative methods for evaluation of dynamic changes in QT interval on 24 hour Holter ECG recordings: QT interval in idiopathic ventricular fi brillation and long QT syndrome. Heart. 2006;92:201–7.
28. Alders M, Koopmann TT, Christiaans I, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fi brillation. Am J Hum Genet. 2009;84:468–76.
29. Allan WC, Timothy K, Vincent GM, Palomaki GE, Neveux LM, Haddow JE. Long QT syndrome in children: the value of rate corrected QT interval and DNA analysis as screening tests in the general population. J Med Screen. 2001;8:173–7.
30. Ghosh S, Cooper DH, Vijayakumar R, et al. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm. 2010;7:534–7.
31. Viskin S, Rosso R, Halkin A. Making sense of early repolarization. Heart Rhythm. 2012;9:566–9.
32. Watanabe H, Nogami A, Ohkubo K, et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fi brillation associated with early repolarization. Circ Arrhythm Electrophysiol. 2011;4:874–81.
33. Belhassen B, Viskin S. Idiopathic ventricular tachycardia and fi brillation. J Cardiovasc Electrophysiol. 1993;4:356–68.
34. Haissaguerre M, Shah DC, Jais P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fi brillation. Lancet. 2002;359:677–8.
35. Haissaguerre M, Shoda M, Jais P, et al. Mapping and ablation of idiopathic ventricular fi brillation. Circulation. 2002;106:962–7.
36. Sinha AM, Schmidt M, Marschang H, et al. Role of left ventricular scar and Purkinje-like potentials during mapping and ablation of ventricular fibrillation in dilated cardiomyopathy. Pacing Clin Electrophysiol. 2009;32:286–90.
37. Arnar DO, Bullinga JR, Martins JB. Role of the Purkinje system in spontaneous ventricular tachycardia during acute ischemia in a canine model. Circulation. 1997;96:2421–9.
38. Ideker RE, Kong W, Pogwizd S. Purkinje fi bers and arrhythmias. Pacing Clin Electrophysiol. 2009;32:283–5.
39. Nogami A. Purkinje-related arrhythmias part ii: polymorphic ventricular tachycardia and ventricular fibrillation. Pacing Clin Electrophysiol. 2011;34:1034–49.
40. Berenfeld O, Jalife J. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3 dimensional model of the ventricles. Circ Res. 1998;82:1063–77.
41. Burashnikov A, Antzelevitch C. Late-phase 3 EAD. A unique mechanism contributing to initiation of atrial fi brillation. Pacing Clin Electrophysiol. 2006;29:290–5.
42. Antzelevitch C, Bernstein MJ, Feldman HN, Moe GK. Parasystole, reentry, and tachycardia: a canine preparation of cardiac arrhythmias occurring across inexcitable segments of tissue. Circulation. 1983;68:1101–15.
43. Viskin S, Belhassen B. Polymorphic ventricular tachyarrhythmias in the absence of organic heart disease. Classification, differential diagnosis and implications for therapy. Prog Cardiovasc Dis. 1998;41:17–34.
44. Haissaguerre M, Sacher F, Nogami A, et al. Characteristics of recurrent ventricular fi brillation associated with inferolateral early repolarization role of drug therapy. J Am Coll Cardiol. 2009;53:612–19.
45. Pasquie JL, Sanders P, Hocini M, et al. Fever as a precipitant of idiopathic ventricular fibrillation in patients with normal hearts. J Cardiovasc Electrophysiol. 2004;15:1271–6.
46. Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long-QT syndrome. Circulation. 1998;98:1928–36.
47. Castro Hevia J, Antzelevitch C, Tornes Barzaga F, et al. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol. 2006;47:1828–34.
48. Abe A, Ikeda T, Tsukada T, et al. Circadian variation of late potentials in idiopathic ventricular fibrillation associated with J waves: insights into alternative pathophysiology and risk strati fi cation. Heart Rhythm. 2010;7:675 82.
49. Nam GB, Ko KH, Kim J, et al. Mode of onset of ventricular fi brillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J. 2009;31:330–9.
50. Rosso R, Glikson E, Belhassen B, et al. Distinguishing “benign” from “malignant early repolarization”: The value of the ST-segment morphology. Heart Rhythm. 2012;9(2):225–9.
51. Champagne J, Geelen P, Philippon F, Brugada P. Recurrent cardiac events in patients with idiopathic ventricular fibrillation, excluding patients with the Brugada syndrome. BMC Med. 2005;3:1.
52. Belhassen B, Pelleg A, Miller HI, Laniado S. Serial electrophysiological studies in a young patient with recurrent ventricular fi brillation. PACE. 1981;4:92–9.
53. Belhassen B, Viskin S, Fish R, Glick A, Setbon I, Eldar M. Effects of electrophysiologic-guided therapy with Class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fi brillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:1301–12.
54. Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494–9.
55. Brugada P, Geelen P, Brugada R, Mont L, Brugada J. Prognostic value of electrophysiologic investigations in Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:1004–7.
56. Shimada M, Miyazaki T, Miyoshi S, et al. Sustained monomorphic ventricular tachycardia in a patient with Brugada syndrome. Jpn Circ J. 1996;60:364–70.
57. Viskin S, Belhassen B. Clinical problem solving: when you only live twice. N Engl J Med. 1995;
332:1221–5.
58. Boersma LV, Jaarsma W, Jessurun ER, Van Hemel NH, Wever EF. Brugada syndrome: a case report of monomorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2001;24:112–15.
59. Dinckal MH, Davutoglu V, Akdemir I, Soydinc S, Kirilmaz A, Aksoy M. Incessant monomorphic ventricular tachycardia during febrile illness in a patient with Brugada syndrome: fatal electrical storm. Europace. 2003;5:257–61.
60. Mok NS, Chan NY, Chiu AC. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol. 2004;27:821–3.
61. Brugada P, Green M, Abdollah H, Wellens HJJ. Signi fi cance of ventricular arrhythmias initiated by programmed ventricular stimulation: the importance of the type of ventricular arrhythmia induced and the number of premature stimuli required. Circulation. 1984;69:87–92.
62. Morady F, DiCarlo L, Baerman J, de Buitleir M. Comparison of coupling intervals that induce clinical and nonclinical forms of ventricular tachycardia during programmed stimulation. Am J Cardiol. 1986;57:1269–73.
63. Stevenson WG, Brugada P, Waldecker B, Zehender M, Wellens HJ. Can potentially signi fi cant polymorphic ventricular arrhythmias initiated by programmed stimulation be distinguished from those that are nonspeci fi c? Am Heart J. 1986;111:1073–80.
64. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: a cardiac ticking time-bomb? Europace. 2007;9:707 10.
65. Viskin S, Rosso R, Rogowski O, Belhassen B. The “short-coupled” variant of right ventricular out fl ow ventricular tachycardia: a not-so-benign form of benign ventricular tachycardia? J Cardiovasc Electrophysiol. 2005;16:912–16.
66. Noda T, Shimizu W, Taguchi A, et al. Malignant entity of idiopathic ventricular fi brillation and polymorphic ventricular tachycardia initiated by premature extrasystoles originating from the right ventricular out fl ow tract. J Am Coll Cardiol.2005;46:1288–94.
67. Myerburg R, Kessler K, Mallon S, et al. Lifethreateningventricular arrhythmias in patientswith silent myocardialischemia due to coronaryarteryspasm. N Engl J Med. 1992;326:1451–5.
68. Wolfe CL, Nibbley C, Bhandari A, Chatterjee K,Scheinman MM. Polymorphous ventriculartachycardia associatedwith acute myocardialinfarction. Circulation. 1991;84:1543–51.
69. Kakishita M, Kurita T, Matsuo K, et al. Mode ofonset of ventricular fi brillation in patients withBrugada syndromedetected by implantable cardioverterde fibrillator therapy. J Am Coll Cardiol.2000;36:1646–53.
70. Chattha IS, Sy RW, Yee R, et al. Utility of the recoveryelectrocardiogram after exercise: a novelindicator for the diagnosis and genotyping oflong QT syndrome? Heart Rhythm. 2010;7:906–11.
71. Krahn AD, Klein GJ, Yee R. Hysteresis of the RTinterval with exercise: a new marker for thelong-QT syndrome? Circulation. 1997;96:1551–6.
72. Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. 2011;124(20):2187–94.
73. Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol. 2010;55:1955–61.
74. Hong K, Brugada J, Oliva A, et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation. 2004;110:3023–7.
75. Wolpert C, Echternach C, Veltmann C, et al. Intravenous drug challenge using fl ecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2:254–60.
76. Ackerman MJ, Khositseth A, Tester DJ, Hejlik JB, Shen WK, Porter CB. Epinephrine-induced QT interval prolongation: a gene-speci fi c paradoxical response in congenital long QT syndrome. Mayo Clin Proc. 2002;77:413 21.
77. Shimizu W, Noda T, Takaki H, et al. Epinephrine unmasks latent mutation carriers with LQT1 form of congenital long-QT syndrome. J Am Coll Cardiol. 2003;41:633–42.
78. Garratt CJ, Antoniou A, Griffith MJ, Ward DE, Camm AJ. Use of intravenous adenosine in sinus rhythm as a diagnostic test for latent preexcitation. Am J Cardiol. 1990;65:868–73.
79. Varnava AM, Elliott PM, Baboonian C, Davison F, Davies MJ, McKenna WJ. Hypertrophic cardiomyopathy: histopathological features of sudden death in cardiac troponin T disease. Circulation. 2001;104:1380–4.
80. Varnava A, Baboonian C, Davison F, et al. A new mutation of the cardiac troponin T gene causing familial hypertrophic cardiomyopathy without left ventricular hypertrophy. Heart. 1999;82:621–4.
81. Fontaine G, Fornes P, Herbert JL. Ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathies. In: Zipes DP, Jalife J, editors. Cardiac electrophysiology: from cell to bedside. 3rd ed. Philadelphia: W.B. Saunders; 2003.
82. Deantonio HJ, Kaul S, Lerman BB. Reversible myocardial depression in survivors of cardiac arrest. Pacing Clin Electrophysiol. 1990;13: 982–5.
83. Viskin S, Halkin A, Olgin JE. Treatable causes of sudden death: not really “treatable” or not really the cause? J Am Coll Cardiol. 2001;38:1725–7.
84. Brembilla-Perrot B, Terrier de la Chaise A, Isaaz K, Marcon F, Cherrier F, Pernot C. Inducible multiform ventricular tachycardia in Wolff-Parkinson- White syndrome. Br Heart J. 1987;58:89–95.
85. Wang Y, Grif fi n J, Lesh M, Cohen T, Chien W, Scheinman M. Patients with supraventricular tachycardia presenting with aborted sudden death: incidence, mechanism and long-term follow- up. J Am Coll Cardiol. 1991;18:1720–1.
86. Tester DJ, Kopplin LJ, Will ML, Ackerman MJ. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005;2:1099–105.
87. Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426–34.
88. Nof E, Belhassen B, Arad M, et al. Postpacing abnormal repolarization in catecholaminergic polymorphic ventricular tachycardia associated with a mutation in the cardiac ryanodine receptor gene. Heart Rhythm. 2011;8:1546–52.
89. Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long QT syndrome. N Engl J Med. 1992;327:846–52.
90. Hong K, Bjerregaard P, Gussak I, Brugada R. Short QT syndrome and atrial fi brillation caused by mutation in KCNH2. J Cardiovasc Electrophysiol. 2005;16:394–6.
91. Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800–7.
92. Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109: 2394–7.
93. Borggrefe M, Wolpert C, Antzelevitch C, et al. Short QT syndrome. Genotype-phenotype correlations. J Electrocardiol. 2005;38:75–80.
94. Belhassen B. A 25-year control of idiopathic ventricular fibrillation with electrophysiologicguided antiarrhythmic drug therapy. Heart Rhythm. 2004;1:352–4.
95. Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54–8.
96. Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter de fi brillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol. 2003;14:1273–7.
97. Strohmer B, Schernthaner C, Pichler M. T-wave oversensing by an implantable cardioverter de fi brillator after successful ablation of idiopathic ventricular fi brillation. Pacing Clin Electrophysiol. 2006;29:431–5.
98. Remme CA, Wever EF, Wilde AA, Derksen R, Hauer RN. Diagnosis and long-term follow-up of the Brugada syndrome in patients with idiopathic ventricular fi brillation. Eur Heart J. 2001;22:400–9.
99. Shimizu W, Matsuo K, Takagi M, et al. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelvelead electrocardiograms. J Cardiovasc Electrophysiol. 2000;11:396–404.
100. Shimeno K, Takagi M, Maeda K, Tatsumi H, Doi A, Yoshiyama M. Usefulness of multichannel holter ECG recording in the third intercostal space for detecting type 1 brugada ECG: comparison with repeated 12-lead ECGs. J Cardiovasc Electrophysiol. 2009;20(9):1026–31.
101. Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J. 2001;22: 2290–6.
102. Viskin S, Antzelevitch C. The cardiologists worst nightmare sudden death from “benign” ventricular arrhythmias. J Am Coll Cardiol. 2005;46: 1295–7.
103. Shimizu W. Arrhythmias originating from the right ventricular out fl ow tract: how to distinguish “malignant” from “benign”? Heart Rhythm. 2009;6:1507–11.
104. Wever EFD, Hauer RNW, Oomen A, Peters RHJ, Bakker PFA, Robles de Medina EO. Unfavorable outcome in patients with primary electrical disease who survived an episode of ventricular fi brillation. Circulation. 1993;88:1021 9.
105. Moe T. Morgagni-Adams-Stokes attacks caused by transient recurrent ventricular fi brillation in a patient without apparent heart disease. Am Heart J. 1949;37:811–18.
106. Konty F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med. 1990; 227:211–13.
107. Belhassen B, Viskin S. Management of idiopathic ventricular fibrillation: implantable defibrillators? Antiarrhythmic drugs? ANE. 1998;3:125–8.
108. Belhassen B, Glick A, Viskin S. Ef ficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731–7.
109. Belhassen B, Shapira I, Sheps D, Shoshani D, Laniado S. Programmed ventricular stimulation using up to two extrastimuli and repetition of double extrastimulation for induction of ventricular tachycardia: A new highly sensitive and speci fi c protocol. Am J Cardiol. 1990;65: 615–22.
110. Belhassen B, Glick A, Viskin S. Excellent longterm reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fi brillation and Brugada syndrome. Pacing Clin Electrophysiol. 2009;32: 294–301.
111. Haghjoo M, Arya A, Heidari A, Sadr-Ameli MA. Suppression of electrical storm by oral quinidine in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2005;16:674.
112. Marquez MF, Rivera J, Hermosillo AG, et al. Arrhythmic storm responsive to quinidine in a patient with Brugada syndrome and vasovagal syncope. Pacing Clin Electrophysiol. 2005;28: 870–3.
113. Marquez MF, Salica G, Hermosillo AG, et al. Ionic basis of pharmacological therapy in Brugada syndrome. J Cardiovasc Electrophysiol. 2007;18: 234–40.
114. Aizawa Y, Tamura M, Chinushi M, et al. An attempt at electrical catheter ablation of the arrhythmogenic area in idiopathic ventricular fibrillation. Am Heart J. 1992;123:257–60.
115. Kusano KF, Yamamoto M, Emori T, Morita H, Ohe T. Successful catheter ablation in a patient with polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol. 2000;11:682–5.
116. Knecht S, Sacher F, Wright M, et al. Long-term follow-up of idiopathic ventricular fi brillation ablation: a multicenter study. J Am Coll Cardiol. 2009;54:522–8.
117. The Antiarrhythmic Versus Implantable Defibrillators (AVID) Investigators. A comparison of antiarrhythmic-drug therapy with implantable de fi brillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med. 1997;337:1576–83.
118. Kron J, Herre J, Renfroe EG, et al. Lead- and device-related complications in the antiarrhythmics versus implantable de fi brillators trial. Am Heart J. 2001;141:92–8.
119. Sacher F, Probst V, Iesaka Y, et al. Outcome after implantation of a cardioverter-de fi brillator in patients with Brugada syndrome: a multicenter study. Circulation. 2006;114:2317–24.


{kind=link}