

TS PHẠM HỮU VĂN
7.4. Bệnh cơ tim phì đại (HCM)
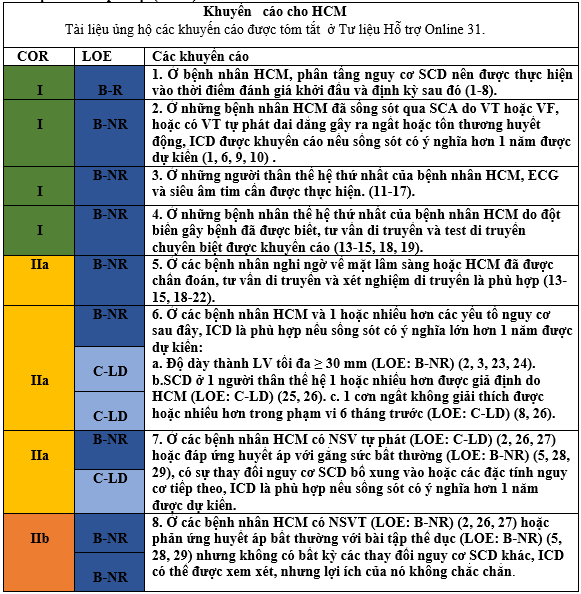
Bảng 8 và hình 7
Tham khảo hướng dẫn của ACCF/AHA về HCM cho khái niệm HCM (36).
Văn bản Hỗ trợ Đặc biệt – Khuyến cáo
1. Bệnh nhân HCM có khoảng 1% nguy cơ SCD mỗi năm (1, 6). Lựa chọn bệnh nhân là ứng viên thích hợp cho cấy ICD có thể là một quyết định lâm sàng khó khăn do tính cả thể của mỗi bệnh nhân và gia đình, các định nghĩa các yếu tố nguy cơ thay đổi và sự thay đổi nhỏ nguy cơ, dữ liệu lâm sàng ít ỏi, sự không phổ biến tương đối của cả hai HCM và SCD trong đại đa số lâm sàng, và các biến chứng tiềm ẩn của đời sống với ICD. Bảng 8 liệt kê các yếu tố nguy cơ và các thay đổi nhỏ của nguy cơ kết hợp với SCD ở các bệnh nhân HCM. Phân tầng nguy cơ ICD nên được thực hiện mỗi 1 đến 3 năm ở bệnh nhân HCM. Có bằng chứng ngày càng tăng lên ủng hộ sự kết hợp của gia tăng gadolium trễ trên MRI tim với nguy cơ đột tử và nó được bao gồm sự biến đổi nhỏ nguy cơ (37-39). Phình thất trái (LV aneurysm) có thể liên quan đến nguy cơ VT đơn hình dai dẳng (40). Tuổi cũng là một xem xét quan trọng, vì nguy cơ đột tử lớn hơn ở những người <30 tuổi, và thấp ở những bệnh nhân có biểu hiện khởi đầu sau 60 tuổi (5, 26), (41).
2. HCM là nguyên nhân phổ biến nhất gây SCD ở các cá thể < 40 tuổi (26). Những cá thể sống sót sau cơn SCD, VF hoặc VT dai dẳng gây ra ngất hoặc tổn thương huyết động chứng minh cho việc cấy ICD (1, 6, 9, 10). Mặc dù không có RCTs đánh giá việc sử dụng ICD ở bệnh nhân HCM có SCD, 1 nghiên cứu cho thấy 54% bệnh nhân có ICD dự phòng đã được ICD điều trị thích hợp trong thời gian theo dõi trung bình 4,6 năm (10 ). Chọn bệnh nhân với HCM có thể là ứng viên cho cấy ICD dưới da (42); tuy nhiên, nhiều dữ liệu hơn về nhóm này là cần thiết đặc biệt do nguy cơ sóng T cao hơn quá mức có thể tăng nguy cơ sốc ICD không phù hợp.
3. Kiểm tra lâm sàng và / hoặc di truyền các thành viên thế hệ thứ nhấ và hai trong gia đình bệnh nhân HCM là rất quan trọng để xác định những người có bệnh không được nhận biết. Tư vấn di truyền cần đi trước test di truyền các thành viên gia đình để gia tăng hiểu biết về tính hữu ích và chi phí xét nghiệm (18, 20, 43). Trên cơ sở bệnh sử gia đình, sàng lọc lâm sàng và phân tích phả hệ, mô hình thừa kế được xác định chắc chắn để xác định và quản lý người thân có nguy cơ (13, 14, 18, 19, 43-45). Do gia đình HCM là rối loạn chiếm ưu thế, nguy cơ một bệnh nhân bị ảnh hưởng sẽ truyền bệnh cho mỗi một người con là 50%. Khi một đột biến gây bệnh được xác định bệnh nhân được lấy ra, tình trạng di truyền của mỗi thành viên trong gia đình có thể được xác định chắc chắn. Những người có quan hệ với HCM rõ ràng sẽ có đột biến HCM bệnh lý tương tự như bệnh nhân được lấy ra. Các đột biến gây bệnh cũng có thể được nhận diện ở các người thân khác có tình trạng lâm sàng chưa biết. Các cá thể đột biến này cần được đánh giá bằng khám thực thể, điện tim đồ (11, 17), và siêu âm tim (12, 16, 17) và, nếu xác định được HCM, những người này phải trải qua quá trình phân tầng nguy cơ. Các đối tượng dương tính không có bằng chứng của HCM có thể HCM có nguy cơ phát triển trong tương lai và được đánh giá lâm sàng (15, 46, 47). Nếu đột biến liên quan đến người được xét nghiệm là đột biến gây bệnh rõ ràng, do các thành viên gia đình đột biến âm tính và con cháu của họ sẽ không có nguy cơ tăng lên phát triển HCM và không cần phải đánh giá tiếp theo. Tuy nhiên, những thành viên trong gia đình đột biến âm tính như vậy cần có bản siêu âm để đảm bảo tính đồng nhất giữa kiểu gene và kiểu hình.
4. Trong một nghiên cứu của 1.053 bệnh nhân có HCM biểu hiện lâm sàng không liên quan, 359 bệnh nhân (34%) có gen dương tính với đột biến liên kết với HCM ở ≥ 1 gen liên kết với HCM (22). Kết quả xét nghiệm di truyền ở những người được xét
nghiệm cải thiện kết cục là không chắc chắn có đúng hay không, nhưng nhận biết đột biến có thể giúp cải thiện kết cục là không chắc chắn, nhưng việc xác định đột biến có thể giúp có biết việc tầm soát người thân.
5. Tư vấn di truyền là quan trọng ở bệnh nhân HCM, và xét nghiệm di truyền của họ hàng cũng rất quan trọng ngoại trừ không có người thân thế hệ thứ nhất hoặc thứ hai. Hầu hết HCM là do đột biến chiếm ưu thế autosom trong các gen mã hóa protein sarcomere hoặc các protein liên quan đến sarcomere. Sự có mặt của đột biến gen sarcomere gây bệnh ở bệnh nhân HCM xác định nguy cơ rối loạn chức năng LV và kết cục bất lợi cho dù có liên quan đến các sợi nhỏ cơ tim (13-15, 18, 19, 22). Một đột biến duy nhất ở 1 trong 2 allele (hoặc bản sao) của một gen đủ để gây ra HCM; Tuy nhiên, 5% bệnh nhân với HCM có ≥ 2 đột biến trong cùng một gen hoặc các gen khác nhau, có thể là một dấu hiệu cho kết quả xấu hơn (13, 34, 48). Khi xét nghiệm di truyền cho thấy một đột biến trong bệnh nhân đưa ra, việc xác định tình trạng di truyền ở những người thân thế hệ thứ nhất và thứ hai có thể tiên đoán nguy cơ phát triển thành HCM (14, 49). Những người có liên quan với HCM sẽ có cùng đột biến bệnh lý như bệnh nhân đưa ra.
6. Một số nghiên cứu đã mô tả mối quan hệ độc lập giữa phì đại và SCD khi mức độ phì đại ≥ 30 mm (2, 3, 23, 24). Nguy cơ không tăng một cách đột ngột đối với các bệnh nhân có độ dầy thành ≥ 30 mm, nhưng tăng lên hơn theo tuyến tính (24) và dường như mang ý nghĩa tiên lượng hơn ở bệnh nhân trẻ tuổi hơn. Người lớn trẻ có phì đại đến 30mm có thể có nguy cơ SCD tương tự hoặc cao hơn bệnh nhân lớn tuổi có độ dày tối đa ≥ 30mm (23, 50).
Bệnh nhân HCM có nguy cơ tăng lên cho SCD nếu họ có một người thân thế hệ thứ nhất đã bị SCD gây ra được giả định do HCM. Bệnh sử gia đình dường như là một dự báo độc lập của SCD mặc dù các nghiên cứu hỗ trợ còn nhỏ và là quan sát (25, 26). Ngất xỉu có thể qua trung gian thần kinh hoặc có liên quan đến thuốc cũng như do VA và đòi hỏi đánh giá cẩn thận trước khi xem xét nó là một yếu tố nguy cơ SCD (8, 26). Trong một phân tích, ngất không giải thích được hoặc không phải qua trung gian thần kinh được kết hợp với nguy cơ SCD chỉ khi nó xuất hiện trong phạm vi 6 tháng nhưng không nếu phần lớn các cơn chỉ xuất hiện > 5 năm trước đó (8).
7. Mặc dù VT kéo dài liên quan chặt chẽ với SCD, dữ liệu đối với NSVT ít mạnh mẽ hơn. Hầu hết các nghiên cứu không ủng hộ NSVT như một yếu tố nguy cơ độc lập cho SCD ở bệnh nhân HCM (2, 26, 27), nhưng nguy cơ tăng lên nếu có các yếu tố nguy cơ nhỏ, đặc biệt ở bệnh nhân dưới 30 tuổi (27). Có tới một phần ba bệnh nhân ở HCM có đáp ứng huyết áp bất thường trong quá trình gắng sức (được định nghĩa hoăc giảm huyết áp 20 mmHg hoặc không tăng huyết áp tâm thu ít nhất 20 mm Hg trong quá trình gắng sức) (28, 29). Phát hiện này đã được giả định là một yếu tố nguy cơ SCD; tuy nhiên, không rõ ràng như thế nào liên quan đến sự gia tăng tắc nghẽn đường ra LV động học xảy ra với gắng sức, trạng thái huyết động có thể dễ dàng thay đổi bằng thuốc hoặc thủ thuật cơ học. Tầm quan trọng của đáp ứng huyết áp bất thường với gắng sức dự đoán nguy cơ SCD tăng lên khi có các nguy cơ hỗ trợ (Bảng 8).
8. Hầu hết các nghiên cứu đều nhận định NSVT đơn thuần có giá trị tiên đoán dương tính thấp cho SCD (2, 26, 27); do đó, việc sử dụng ICD sẽ phù hợp hơn nếu có các các yếu tố nguy cơ hỗ trợ. Một đáp ứng huyết áp bất thường đối với gắng sức cũng có liên quan đến nguy cơ đột tử (5, 28, 29), nhưng không rõ ràng điều này liên quan như thế nào đối với sự tăng lên trong tắc nghẽn đường ra thất trái về huyết động xảy ra với gắng sức, thường có thể điều trị được. Tầm quan trọng của đáp ứng huyết áp bất thường với gắng sức để dự đoán nguy cơ SCD tăng lên khi có các yếu tố nguy cơ hỗ trợ (Bảng 8).
8. Hầu hết các nghiên cứu đều nhận định NSVT đơn thuần có giá trị tiên đoán dương tính thấp cho SCD (2, 26, 27); do đó, việc sử dụng ICD sẽ phù hợp hơn nếu có các các yếu tố nguy cơ hỗ trợ. Đáp ứng huyết áp bất thường đối với gắng sức cũng có liên quan đến nguy cơ đột tử (5, 28, 29), nhưng không rõ ràng điều này liên quan như thế nào đối với sự tăng lên trong tắc nghẽn đường ra thất trái về huyết động xảy ra với gắng sức, thường có thể điều trị được. Tầm quan trọng của đáp ứng huyết áp bất thường với gắng sức để dự đoán nguy cơ SCD tăng lên khi có các yếu tố nguy cơ hỗ trợ (Bảng 8).
9. ICD được khuyến cáo để phòng ngừa SCD ở các bệnh nhân HCM đã sống sót do VT hoặc VF khi các thuốc chống loạn nhịp có hiệu quả hạn chế (31). Amiodaron có liên quan đến sống sót được cải thiện trong các nghiên cứu quan sát và là một lựa chọn cho các bệnh nhân khi ICD không khả thi do hy vọng sống sót bị hạn chế hoặc ưa thích của bệnh nhân (30, 31).
10. Khoảng một phần ba số bệnh nhân HCM liên tiếp trải qua nghiên cứu điện sinh lý có VT đa hình hoặc VF được tạo ra bằng kích thích thất có chương trình, nhưng các kết quả kích thích có chương trình không dự bào được nguy cơ SCD. Kích thích thất có chương trình ở bệnh nhân HCM có giá trị tiên đoán thấp và nguy cơ biến chứng đáng kể (32, 33, 51). Các nghiên cứu điện sinh lý có thể giúp làm rõ chẩn đoán nhịp tim nhanh phức hợp rộng hoặc hướng dẫn điều trị nhịp tim nhanh trên thất hoặc vào lại nhánh bó.
Bảng 8. Các đặc tính lâm sàng lớn được kết hợp với nguy cơ tăng lên của SCD ở Bệnh nhân HCM

* Có sự đồng thuận chung trong tài liệu các yếu tố này làm nguy cơ gia tăng một cách độc lập cho SCD ở bệnh nhân HCM.
† Giảm huyết áp 20 mm Hg hoặc không tăng huyết áp tâm thu > 20 mm Hg trong quá trình gắng sức.
‡ Không có sự đồng thuận trong tài liệu những yếu tố hỗ trợ này có thể làm nguy cơ SCD tăng lên một cách độc lập ở các bệnh nhân HCM; tuy nhiên, các yếu tố hỗ trợ nguy cơ khi kết hợp với yếu tố nguy cơ thường xác định bệnh nhân với HCM có nguy cơ SCD tăng lên ngoài nguy cơ đã được chuyền đạt bằng yếu tố nguy cơ đơn độc.
§ Một nhóm nhỏ các bệnh nhân có LVEF < 50% ( bệnh giai đoạn cuối) hoặc phình LV đảm bảo xem xét cho việc cấy ICD (52).
HCM: bệnh cơ tim phì đại; ICD: máy khử rung tim có thể cấy; LV: thất trái; LVEF: phân suất tống máu thất trái; LVOT: đường ra thất trái; NSVT: nhịp nhanh thất tạm thời; SCD: đột tử tim; VT: nhịp nhanh thất; và VF: rung tâm thất.
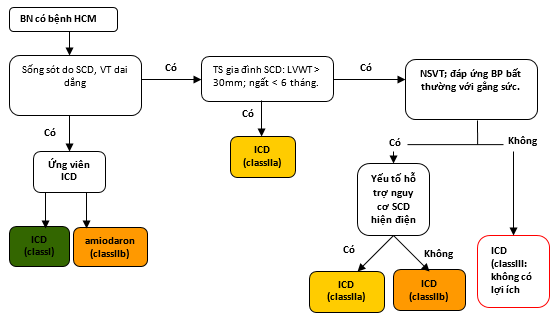
Hình 7. Ngăn ngừa SCD ở các Bệnh nhân HCM
Các màu tương ứng với Class của các khuyến cáo ở bảng 1.
Xem phần 7.4 cho thảo luận.
* Ứng viên ICD được xác định theo tình trạng chức năng, tuổi thọ, hoặc sở thích của bệnh nhân.
† Các yếu tố hỗ trợ nguy cơ: Tuổi < 30, gia tăng gadolinium trễ trên MRI tim, tắc nghẽn LVOT, phình LV, ngất > 5 năm. BP: huyết áp; HCM: bệnh cơ tim phì đại; Hx: bệnh sử; ICD: máy khử rung tim có thể cấy; LVOT: đường ra thất trái; LVWT: độ dày vách thất trái; MRI: hình ảnh cộng hưởng từ; NSVT: nhịp nhanh thất tạm thời; SCD: đột tử tim; và VT: nhịp nhanh thất.
Tài liệu tham khảo
1. Elliott PM, Sharma S, Varnava A, et al. Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999;33:1596-601.
2. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-8.
3. Elliott PM, Gimeno Blanes JR, Mahon NG, et al. Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet. 2001;357:420-4.
4. Elliott PM, Gimeno JR, Tome MT, et al. Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J. 2006;27:1933-41.
5. Maki S, Ikeda H, Muro A, et al. Predictors of sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol. 1998;82:774-8.
6. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-12.
7. O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014;35:2010-20.
8. Spirito P, Autore C, Rapezzi C, et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703-10.
9. O’Mahony C, Lambiase PD, Quarta G, et al. The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophic cardiomyopathy. Heart. 2012;98:116-25.
10.Syska P, Przybylski A, Chojnowska L, et al. Implantable cardioverter-defibrillator in patients with hypertrophic cardiomyopathy: efficacy and complications of the therapy in long-term follow-up. J Cardiovasc Electrophysiol. 2010;21:883-9.
11.Adabag AS, Kuskowski MA, Maron BJ. Determinants for clinical diagnosis of hypertrophic cardiomyopathy. Am J Cardiol. 2006;98:1507-11.
12.Afonso LC, Bernal J, Bax JJ, et al. Echocardiography in hypertrophic cardiomyopathy: the role of conventional and emerging technologies. JACC Cardiovasc Imag. 2008;1:787-800.
13.Girolami F, Ho CY, Semsarian C, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444-53.
14.Ingles J, Sarina T, Yeates L, et al. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy. Genet Med. 2013;15:972-7.
15.Jensen MK, Havndrup O, Christiansen M, et al. Penetrance of hypertrophic cardiomyopathy in children and adolescents: a 12-year follow-up study of clinical screening and predictive genetic testing. Circulation. 2013;127:48-54.
16.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26:1699-708.
17.Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy. Clinical spectrum and treatment. Circulation. 1995;92:1680-92.
18.Christiaans I, Birnie E, van Langen IM, et al. The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: focus on predictive screening. Eur Heart J. 2010;31:842-8.
19.Olivotto I, Girolami F, Ackerman MJ, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83:630-8.
20.Christiaans I, van Langen IM, Birnie E, et al. Genetic counseling and cardiac care in predictively tested hypertrophic cardiomyopathy mutation carriers: the patients’ perspective. Am J Med Genet. 2009;149a:1444-51.
21.Hamang A, Eide GE, Rokne B, et al. Predictors of heart-focused anxiety in patients undergoing genetic investigation and counseling of long QT syndrome or hypertrophic cardiomyopathy: a one year follow-up. J Genet Counsel. 2012;21:72-84.
22.Bos JM, Will ML, Gersh BJ, et al. Characterization of a kiểu hình-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89:727-37.
23.Sorajja P, Nishimura RA, Ommen SR, et al. Use of echocardiography in patients with hypertrophic cardiomyopathy: clinical implications of massive hypertrophy. J Am Soc Echocardiogr. 2006;19:788-95.
24.Spirito P, Bellone P, Harris KM, et al. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778-85.
25.Bos JM, Maron BJ, Ackerman MJ, et al. Role of family history of sudden death in risk stratification and prevention of sudden death with implantable defibrillators in hypertrophic cardiomyopathy. Am J Cardiol. 2010;106:1481-6.
26.Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;342:365-73.
27.Monserrat L, Elliott PM, Gimeno JR, et al. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol. 2003;42:873-9.
28.Olivotto I, Maron BJ, Montereggi A, et al. Prognostic value of systemic blood pressure response during exercise in a community-based patient population with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1999;33:2044-51.
29.Sadoul N, Prasad K, Elliott PM, et al. Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation. 1997;96:2987-91.
30.McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-6.
31.Melacini P, Maron BJ, Bobbo F, et al. Evidence that pharmacological strategies lack efficacy for the prevention of sudden death in hypertrophic cardiomyopathy. Heart. 2007;93:708-10.
32.Kuck KH, Kunze KP, Schluter M, et al. Programmed electrical stimulation in hypertrophic cardiomyopathy. Results in patients with and without cardiac arrest or syncope. Eur Heart J. 1988;9:177-85.
33.Zhu DW, Sun H, Hill R, et al. The value of electrophysiology study and prophylactic implantation of cardioverter defibrillator in patients with hypertrophic cardiomyopathy. Pacing Clin Electrophysiol. 1998;21:299-302.
34.Ackerman MJ, VanDriest SL, Ommen SR, et al. Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002;39:2042-8.
35.Lopes LR, Rahman MS, Elliott PM. A systematic review and meta-analysis of kiểu gene-kiểu hình associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart. 2013;99:1800-11.
36.Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation. 2011;124:783-831.
37.Green JJ, Berger JS, Kramer CM, et al. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2012;5:370-7.
38.O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:867-74.
39.Rubinshtein R, Glockner JF, Ommen SR, et al. Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3:51-8.
40.Rowin EJ, Maron BJ, Haas TS, et al. Hypertrophic cardiomyopathy with left ventricular apical aneurysm: implications for risk stratification and management. J Am Coll Cardiol. 2017;69:761-73.
41.Maron BJ, Rowin EJ, Casey SA, et al. Risk stratification and outcome of patients with hypertrophic cardiomyopathy >=60 years of age. Circulation. 2013;127:585-93.
42.Francia P, Adduci C, Palano F, et al. Eligibility for the subcutaneous implantable cardioverter-defibrillator in patients with hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol. 2015;26:893-9.
43.Christiaans I, Birnie E, Bonsel GJ, et al. Manifest disease, risk factors for sudden cardiac death, and cardiac events in a large nationwide cohort of predictively tested hypertrophic cardiomyopathy mutation carriers: determining the best cardiological screening strategy. Eur Heart J. 2011;32:1161-70.
44.Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899-908.
45.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248-57.
46.Maron BJ, Semsarian C. Emergence of gene mutation carriers and the expanding disease spectrum of hypertrophic cardiomyopathy. Eur Heart J. 2010;31:1551-3.
47.Maron BJ, Yeates L, Semsarian C. Clinical challenges of kiểu gene positive (+)-kiểu hình negative (-) family members in hypertrophic cardiomyopathy. Am J Cardiol. 2011;107:604-8.
48.Ingles J, Doolan A, Chiu C, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42:e59.
49.Rosenzweig A, Watkins H, Hwang DS, et al. Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med. 1991;325:1753-60.
50.O’Mahony C, Jichi F, Monserrat L, et al. Inverted U-shaped relation between the risk of sudden cardiac death and maximal left ventricular wall thickness in hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol. 2016;9:e003818.
51.Saeed M, Homoud MK, Wang PJ, et al. Role of invasive electrophysiologic testing in risk stratification for sudden cardiac death. J Invasive Cardiol. 2001;13:758-62.
52.Maron BJ, Haas TS, Goodman JS. Hypertrophic cardiomyopathy: one gene … but many kiểu hìnhs. Am J Cardiol. 2014;113:1772-3.
53.Ho CY, Cirino AL, Lakdawala NK, et al. Evolution of hypertrophic cardiomyopathy in sarcomere mutation carriers. Heart. 2016;102:1805-12.
54.Moon JC, McKenna WJ, McCrohon JA, et al. Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol. 2003;41:1561-7.
55.Maron MS, Finley JJ, Bos JM, et al. Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. 2008;118:1541-9.
56.Minami Y, Kajimoto K, Terajima Y, et al. Clinical implications of midventricular obstruction in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2011;57:2346-55.


{kind=link}