

Định nghĩa và phân loại tăng áp phổi và tăng áp động mạch phổi
1. Định nghĩa: Tăng áp phổi (pulmonary hypertension: PH) được định nghĩa khi áp lực động mạch phổi trung bình (mPAP) ≥ 25mmHg được đo bằng thông tim phải lúc nghỉ ngơi 1.
TS. BS. Trần Văn Hùng
Bệnh viện – Trường Đại học Kobe, Nhật Bản
Bình thường mPAP là 14± 3,3 mmHg2. Tại hội nghị thế giới về PH lần thứ 4 (Dana Point, California 2008), PH được chia làm 5 nhóm theo bệnh căn và phân loại này được cập nhật tại hội nghị lần thứ 5 (Nice, Pháp 2013) (Bảng 1)3, 4. Nhóm 1 được xem là tăng áp động mạch phổi (pulmonary arterial hypertension: PAH), trong khi đó 4 nhóm còn lại được xem như là PH.
Bảng 1: Cập nhật phân loại lâm sàng tăng áp phổi (PH) (Nice, 2013)
|
Nhóm 1: Tăng áp động mạch phổi (PAH) 1.1 PAH vô căn 1.2 PAH liên quan yếu tố di truyền 1.3 PAH do thuốc hay độc chất 1.4 PAH liên quan với: 1.4.1 Bệnh mô liên kết 1.4.2 Nhiễm HIV 1.4.3 Tăng áp tĩnh mạch cửa 1.4.4 Bệnh tim bẩm sinh 1.4.5 Nhiễm Schistosomiasis Nhóm 1’: Bệnh tắc nghẽn tĩnh mạch phổi và/hoặc u mạch máu maomạch phổi (pulmonary capillary hemangiomatosis) Nhóm 1’’: Tăng áp phổi trường diễn ở trẻ sơ sinh Nhóm 2: Tăng áp phổi (PH) do tim trái 2.1 Rối loạn chức năng tâm thu thất trái. 2.2 Rối loạn chức năng tâm trương thất trái 2.3 Bệnh van tim 2.4 Tắc nghẽn đường vào/ra thất trái bẩm sinh hoặc mắc phải và bệnh cơ tim bẩm sinh Nhóm 3: PH do bệnh phổi và/hoặc thiếu oxy 3.1 Bệnh phổi tắc nghẽn mạn tính (COPD) 3.2 Bệnh phổi mô kẽ 3.3 Các bệnh phổi khác với phối hợp hạn chế và tắc nghẽn 3.4 Rối loạn hô hấp khi ngủ 3.5 Rối loạn thông khí phế nang 3.6 Sống lâu ở vùng cao so với mực nước biển(chronic exposure to high altitude) 3.7 Bất thường về phát triển phổi Nhóm 4: PH do huyết khối thuyên tắc mạn tính (CTEPH) Nhóm 5: PH có cơ chế không rõ ràng |
Hiện nay thuốc điều trị đặc hiệu PH (PH-specific therapy) chỉ được Cơ quan Quản lý Dược phẩm và Thực phẩm Hoa Kỳ (FDA) chấp thuận cho điều trị PH nhóm 1 (PAH) và nhóm 4 (CTEPH) (chỉ có một thuốc là riociguat)5. Nhóm 2 là nhóm PH mà người bác sĩ thường gặp nhất trên thực hành lâm sàng ngày nay 6, tuy nhiên cho đến nay vẫn chưa có thuốc điều trị đặc hiệu được chấp thuận.
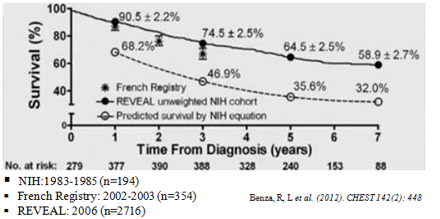
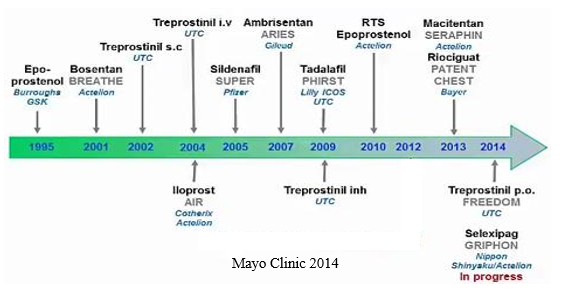
PAH: là một bệnh nặng gây nên bởi sự co mạch và tái cấu trúc mạch máu phổi (có thể có kèm huyết khối) dẫn đến tăng kháng lực mạch máu phổi (PVR) và áp lực động mạch phổi (PAP) làm dày tâm thất phải, cuối cùng là suy tim phải và tử vong7-9. PAH được chẩn đoán khi áp lực trung bình mPAP ≥ 25mmHg và áp lực động mạch phổi bít ≤ 15mmHgđược đo bằng thông tim phải lúc nghỉ ngơi. Mặc dù PAH có bệnh căn từ nhiều nguyên nhân khác nhau (Bảng 1) nhưng cùng có chung đặc điểm bệnh học mà các nhóm PH khác cũng như các bệnh do tuần hoàn hệ thống gây nên không có đó là trong tái cấu trúc mạch máu phổi ngoài sự tăng sinh và phì đại tế bào cơ trơn mạch máu còn có sự tăng sinh của tế bào nội mạc tiểu động mạch phổi và cuối cùng tạo thành thương tổn plexiform10, đây là một thương tổn đặc trưng của PAH giai đoạn cuối và có đặc điểm giống như ung thư 11-13. PAH có tỉ lệ tử vong cao và hiện tại vẫn chưa có thuốc chữa khỏi 14. Tuy nhiên nhờ những tiến bộ trong những năm gần đây trong điều trị và chăm sóc dựa trên những hiểu biết mới về cơ chế sinh bệnh học, phát triển dược phẩm và những thử nghiệm lâm sàng quy mô lớn có độ tin cậy cao mà tỉ lệ sống còn và chất lượng cuộc sống của bệnh nhân PAH tăng lên rõ rệt (Hình 1) 15. Những thuốc điều trị đặc hiệu hiện tại được chấp thuận bởi FDA và Cơ quan Quản lý Dược phẩm Châu Âu (EMA) có cơ chế tác dụng chỉ tập trung vào 3 con đường cơ chế chính: Prostacyclin gồm epoprostenol, illoprost, treprostinil; Nitric oxide (NO) gồm sildenafil, tadalafil, riociguat; Endothelin gồm bosentan, ambrisentan và macitentan.Thời điểm những thuốc trên được FDA chấp thuận cùng với các thử nghiệm lâm sàng của chúngcũng như công ty sản xuất được chỉ ra trong hình 2. Trong bài này chúng tôi chỉ tập trung vào thuốc tác động vào con đường endothelin (nhóm kháng thụ thể endothelin (ERAs)).
Hình 1: So sánh tỉ lệ sống còn của bệnh nhân PAH từ 3 nghiên cứu sổ bộ trước (NIH) và sau khi có điều trị đặc hiệu (French và REVEAL)
Hình 2: Thuốc được FDA chấp thuận cho điều trị PAHtheo thời gian
Sinh lý hệ endothelin
1. Endothelin: là một nhóm peptide có 21 acid amin và đóng vai trò quan trọng trong điều hòa trương lực mạch máu. Endothelin-1 (ET-1) được chiết bởi Yanagisawa và cộng sự (cs) (1988) 16. 2 thành viên còn lại (ET-2 và ET-3) được tìm ra 1 năm sau đó (1989) 17. Endothelin được tiết ra chủ yếu từ tế bào nội mạc mạch máu, tuy nhiên cũng được tiết ra từ những tế bào khác như: tế bào thượng bì phế quản, đại thực bào, tế bào cơ trơn mạch máu, tế bào cơ tim, tế bào trung mô cầu thận và glial cell18-21. ET-2 được tìm thấy nhiều ở thận 22, ET-3 được tìm thấy nhiều ở ruột và não23, 24. Endothelin là chất nội sinh co mạch mạnh và kéo dài 16, 22, 25. Endothelin liên quan đến nhiều tình trạng bệnh gồm ung thư, co thắt phế quản, bệnh xơ hóa, suy timvàPH26-28.
2. Thụ thể endothelin
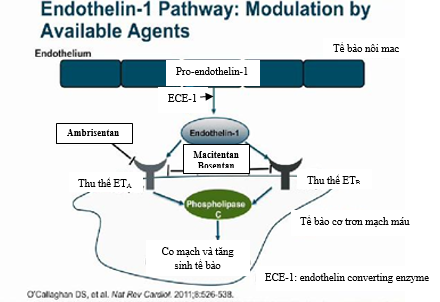
Cả 3 endothelin gắn với 2 thụ thể là ETA và ETB trên bề mặt tế bào.ETA được tìm thấy trên tế bào cơ trơn mạch máu và tế bào cơ tim, trong khi đó ETB nằm cả trên tế bào nội mạc và cơ trơn mạch máu 29. Kích hoạt thụ thể ETA và ETB trên tế bào cơ trơn mạch máu gây nên co mạch mạnh và tăng sinh tế bào30-32. Trái lại khi kích thích thụ thể ETB trên tế bào nội mạc mạch máu đưa đến tăng sản xuất và phóng thích nitric oxide (NO) và prostacyclin từ tế bào nội mạc (là 2 chất dãn mạch mạnh và kháng tăng sinh tế bào)33đồng thời ức chế sản xuất ECE-1 trên tế bào nội mạc 34. ECE-1 là một men ly giải ppET-1 (tiền chất ET-1 không hoạt tính) thành ET-1 peptide có hoạt tính.Phổi người khỏe mạnh sản xuất khoảng 50% lượng ET-1 và cũng góp phần thanh thải 50% lượng này trong tuần hoàn thông qua thụ thể ETB35. Số lượng ETB trên tế bào cơ trơn động mạch phổi tăng lên và tác dụng co mạch của ET-1 thông qua ETBcũng mạnh hơn trên động mạch phổi của cá thểbịPAH so với cá thể bình thường 36, 37. Điều này cũng có thể giải thích tại sao ích lợi của ERAsETA chọn lọc không vượt trội ERAs không chọn lọc trong điều trị PAH38, 39.
Sinh lý bệnh hệ endothelin trên bệnh nhân PAH
Trên bệnh nhân PAH vô căn: nồng độ ET-1 trong huyết tương liên quan đến độ nặng và tiên lượng của bệnh 40, 41, đặc biệt là có sự tăng sản xuất hoặc giảm thanh thải ET-1 từ phổi của những bệnh nhân này được xác định bởi nồng độ ET-1 trong máu động mạch cao hơn so với trong máu tĩnh mạch 41.Đặc biệt ET-1 tăng lên trong nội mạc động mạch phổi và trong thương tổn plexiform 42.
Ở bệnh nhân PAH do bệnh tim bẩm sinh: Nồng độ ET-1 huyết tương tăng do phổi tăng sản xuất và tỉ lệ với áp lực động mạch phổi 43. Nồng độ này giảm về bình thường sau khi phẫu thuật sửa chữa bệnh tim thành công 44. ET-1 và ETA tăng cao trong động mạch phổi 45.
Vai trò ERAs trong điều trị PAH
Hiện tại có 3 thuốc ERAs được FDA chấp thuận cho điều trị PAH gồm: bosentan, ambrisetan và macitentan. Cơ chế tác dụng của chúng được trình bày trong hình3.
2 thuốc đầu tiên được FDA chấp thuận cho điều trị PAH dựa trên kết quả của những thử nghiệm lâm sàng phân nhóm ngẫu nhiên có đối chứng với giả dược, kết quả chỉ ra hiệu quả của chúng trong cải thiện khả năng gắng sức, được xác định bằng thay đổi khoảng cách đi bộ trong 6 phút (∆6MWD) và phân độ chức năng theo WHO (WHO-FC). 2 chỉ số này đã được chứng minh là liên quan chặt chẽ đến tỉ lệ sống còn của bệnh nhân qua các nghiên cứu lâm sàng46, 47. Những nghiên cứu mở nhãn dùng ERAs trong thời gian dài cũng đã xác nhận hiệu quả cũng như an toàn của những thuốc này. Macitentan được chấp thuận dựa trên kết quả nghiên cứu SERAHIN
Hình 3: Cơ chế tác dụng của các thuốc kháng thụ thể endodothelin (ERAs)
1. Ambrisentan
Ambrisentan là một ERA chọn lọc trên thụ thể ETA. Thuốc được xác định hiệu quả từ 2 thử nghiệm lâm sàng phân nhóm ngẫu nhiên, mù đôi, đa trung tâm và có đối chứng (ARIES-1:202 bệnh nhân chia làm 3 nhóm dùng: 5 mg hoặc 10 mg hoặc giả dược; ARIES-2: 192 bệnh nhân dùng: 2.5 mg hoặc 5 mg hoặc giả dược) được thực hiện kéo dài 12 tuần 48. Kết quả cả 2 thử nghiệm điều đạt được tiêu chí chính là tăng∆6MWD có ý nghĩa so với giả dược.Sau 12 tuần điều trị ambrisentan 5 và 10mg làm tăng ∆6MWD tương ứnglà 31 m (p=0,008) và 51 m (p<0.001) trong nghiên cứu ARIES-1; và 32 m (p=0.022) và 59 m (p<0.001) tương ứng liều 2,5 và 5 mg trong nhiên cứu ARIES-2. Đặc biệt, trong nghiên cứu ARIES-2 ambrisentan làm kéo dài thời gian dẫn đến diễn tiến xấu trên lâm sàng (TCW), trong khi đó chỉ số này không cải thiện có ý nghĩa trong nghiên cứu ARIES-1 (p=0.307). Thêm vào đó, WHO-FC cải thiện có ý nghĩa trong nghiên cứu ARIES-1 nhưng không có ý nghĩa trong nghiên cứu ARIES-2 (p=0.117). 298 bệnh nhân từ 2 nghiên cứu trên được tuyển vào 1 nghiên cứu mở rộng kéo dài ít nhất 48 tuần. 18 bệnh nhân phải điều trị phối hợp với prostacyclin hoặc PDE-5 inhibitors. Trong số 280 bệnh nhân tiếp tục đơn trị liệu ambrisentan, thuốc làm tăng ∆6MWD 40 m (KTC95% từ 33 m đến 48 m) sau 12 tuần và 39 m (KTC 95% từ 29 đến 49 m) sau 48 tuần so với lúc mới vào nghiên cứu.Mặc dù không có bệnh nhân nào có men gan (ALT hoặc AST) tăng hơn 3 lần giới hạn trên của bình thường khi điều trị bằng ambrisentan, nhưng khi theo dõi thời gian dài thấy xuất hiện một vài trường hợp tăng men gan nhưng trở về bình thường khi ngưng điều trị.
2. Bosentan
Bosentan là một ERA không chọn lọc, có ái lực mạnh hơn với thụ thểETAvà là thuốc đầu tiên trong nhóm được FDA chấp thuận cho điều trị PAH (2001). Thử nghiệm lâm sàng đầu tiên dùng bosentan là một thử nghiệm lâm sàng phân nhóm ngẫu nhiên, mù đôi và có đối chứng trên 32 bn bị PAH vô căn (84%) hoặc PAH liên quan với xơ cứng bì có NYHA III được dùng bosentan hoặc giả dược trong 12 tuần49. Tiêu chí chính là thay đổi ∆6MWD sau 12 tuần so với giả dược.Tiêu chí phụ là thay đổi huyết động học mạch phổi,WHO-FC. Sau 12 tuần điều trị bosentan làm tăng ∆6MWD 76 m, cùng với cải thiện có ý nghĩa huyết động học mạch phổi, chỉ sốtim và kháng lực mạch máu phổi (PVR). Về tác dụng phụ, tăng men gan không triệu chứng gặp trong 2 bệnh nhândùng bosentan nhưng trở về bình thường sau khi ngưng thuốc hoặc giảm liều.
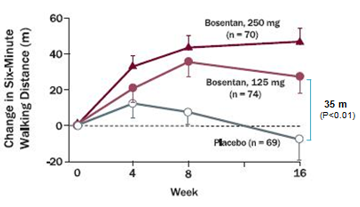
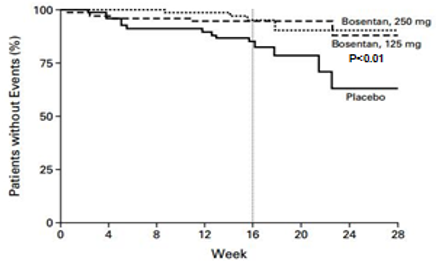
Từ kết quả nghiên cứu trên, Rubin và cs tiến hành mộtthử nghiệm lâm sàng lớn BREATHE-1 (Bosentan randomized trial of endothelin antagonisttherapy) phân nhóm ngẫu nhiên, mù đôi, đa trung tâm và có đối chứng thực hiện trên 213 bệnh nhân50. Bệnh nhân bị PAH vô căn chiếm 70% còn lại là PAH liên quan bệnh mô liên kết, có tuổi từ 12- 80 tuổivà có WHO-FC III-IV, được phân vào 1 trong 3 nhóm: nhóm dùng giả dược, 2 nhóm còn lại dùng bosentan 62,5 mg x 2/ngày trong 4 tuần sau đó tăng lên và duy trì ở liềuhoặc 125 mg x 2/ngày hoặc 250 mg x 2/ngày trong 28 tuần.Tiêu chíchínhlà ∆6MWD sau 16 tuần điều trị. Tiêu chí phụ gồm thay đổi WHO-FC và khoảng thời gian từ khi bắt đầu điều trị đến khi xuất hiện dấu hiệu trở nặng trên lâm sàng (TCW) (dấu hiệu trở nặng trên lâm sàng là phối hợp các biến cố tử vong, ghép phổi, nhập việnvì lý do tăng áp phổi, không cải thiện lâm sàng hoặc diễn tiến xấu dẫn đến ngưng điều trị, phải dùng epoprostenol truyền tĩnh mạch, hoặc cắt vách liên nhĩ). Sau 16 tuần điều trị, bosentan làm tăng ∆6MWD35 m và 54 m có ý nghĩa thống kê tương ứng với mức liều 150mg x 2/ngày và 250 mg x 2/ngày (Hình 4).Ngoài ra bosentan còn làm cải thiện WHO-FC và đặc biệt kéo dài TCW có ý nghĩa thống kê sau 16 tuần và 28 tuần ở cả 2 mức liều điều trị (Hình 5).Tác dụng phụ: liều 125mg x2/ngày không làm tăng có ý nghĩa tác dụng phụ, trong khi liều 250mg x 2/ngày làm tăng men gan so với giả dược. Từ kết quả nghiên cứu này, mặc dầu bosentan có hiệu quả cao hơn với liều cao 250 mg x 2/ngày nhưng do có tác dụng độc trên gan tăng theo liều dùng nên FDA và EMA ủng hộ cho sử dụng liều 125 mg x 2/ngày.
Hình 4: Thay đổi trung bình 6MWD từ lúc khởi trị đến tuần thứ 16 ở2 nhóm dùng bosentan và nhóm giả dược
Hình 5: Tỉ lệ bệnh nhân không xuất hiện dấu hiệu trở nặng trên lâm sàng
Hiệu quả và an toàn của bosentan trên bệnh nhân PAH do bệnh tim bẩm sinh và có hội chứng Eisenmenger
Năm 2006, Canada mở rộng chỉ định điều trị của bosentan cho những bệnh nhân PAH do bệnh bẩm sinh có luồng thuông chủ phổicó WHO-FC III-IV, đồng thời châu Âu cũng chấp thuậnchỉ định ở những bệnh nhân PAH có luồng thông chủ phổi bẩm sinh hoặc hội chứng Eisenmenger. Sự ủng hộ này dựa trên kết quả của nghiên cứu BREATHE-5 được thực hiện bởi Galie và cs 51. Đây là 1 thử nghiệm lâm sàng phân nhóm ngẫu nhiên, mù đôi, đa trung tâm và có đối chứng thực hiện trên 54 bệnh nhânbị hội chứng Eisenmenger do tim bẩm sinh có luồng thuông trái-phải (thông liên thất, thông liên nhĩ và phối hợp),≥ 12 tuổi, WHO-FC III và SpO2lúc nghỉ 70-90% được dùng giả dược hoặc bosentan khởi đầu 62,5 mg x 2/ngày trong 4 tuần sau đótăng lên 125mg x 2/ngày trong 12 tuần. Tiêu chí chính gồm thay đổi SpO2(tiêu chí về tính an toàn) và PVR (tiêu chí về hiệu quả) được đánh giá bằng thông tim phải.Tiêu chí phụ gồm thay đổi mPAP, WHO-FC và ∆6MWD.Kết quả cho thấy SpO2không khác biệt có ý nghĩa thống kê giữa trước điều trị và sau điều trị cũng như giữa nhóm bosentan và giả dược (83.6 ± 5.1% so với 83.7±6.2% và 82.4 ± 53% so với 80.2 ± 8.9% trong nhóm giả dược và nhóm bosentan, theo thứ tự). Bosentan cũng làm giảm PVR có ý nghĩa so với giả dược (p=0,0383) (bảng 2).Với tiêu chí phụ, bosentan cũng làm giảm mPAP (p=0,0363) (bảng 2),tăng ∆6MWD53 m (p=0,008) và cải thiện WHO-FC. Tác dụng phụ thường xảy ra trong nhóm bosentan so với giả dược là phù ngoại biên (19% so với 6%), nhức đầu (14% so với 12%), hồi hộp (11% so với 0%) và tăng men gan (3% so với 0%). Kết luận bosentan an toàn và hiệu quả trên bệnh nhân bị hội chứng Eisenmenger.
Bảng 2: Ảnh hưởng huyết động sau điều trị 16 tuần bằng giả dược và bosentan

Hiệu quả bosentan trên bệnh nhân PAH có triệu chứng nhẹ (WHO-FCII)
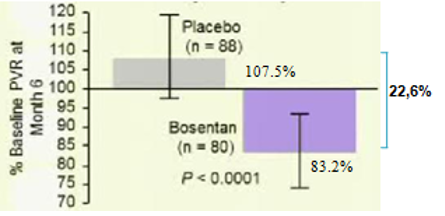
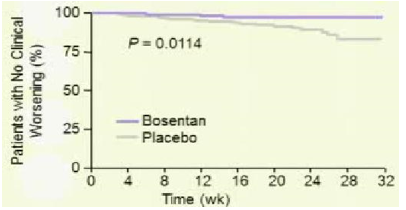
Trước năm 2008, các thuốc điều trị đặc hiệu PAH chỉ được khuyến cáo dùng cho bệnh nhân PAH có WHO-FC III-IV. Tuy nhiên lúc này nhiều câu hỏi được đặt ra là liệu các thuốc điều này có mang lại lợi ích cho bệnh nhân giai đoạn sớm của bệnh hay không? Bosentan là thuốc duy nhất cho đến nay được dùng trong nghiên cứu (EARLY) là thiết kế chỉ ra lợi ích lớn của việc điều trị sớm trên những bệnh nhân có WHOFC II52. Đây là 1 thử nghiệm lâm sàng phân nhóm ngẫu nhiên, mù đôi, đa trung tâm và có đối chứng thực hiện trên 185 bệnh nhân PAH có WHO-FC II, tuổi ≥12 (bảng 3) được dùng bosentan liều khởi đầu 62,5 mg x2/ngày trong 4 tuần và sau đó tăng lên liều đích 125 mg x2/ngày (nếu bệnh nhân có cân nặng< 40kg thì vẫn giữ nguyên liều ban đầu) hoặc giả dược trong 6 tháng. Tiêu chí chính gồm thay đổi PVR và ∆6MWD. Tiêu chí phụ gồm thay đổi WHO-FC, TCW, mPAP và chỉ số tim. Kết quả 6 tháng điều trị cho thấy bosentan làm giảm PVR 22,6% so với giả dược (KTC 95% từ -33,5 đến -10, p<0,0001) (Hình 6) và có khuynh hướng cải thiện ∆6MWD19,1 m (KTC 95% từ -3,6 đến 41,8; p= 0.0758). Bosentanlàm kéo dài TCW (HR 0,227, KTC 95% 0,065-0,798; p=0,0114) (hình 7), giảm số bệnh nhân tăng độ WHO-FC so với giả dược (3 bệnh nhân (3,4%) so với 12 (13,2%)), giảm NT-pro-BNP (KTC 95%từ -749 đến -192; p=0,0003), cải thiện chỉ số tim 0,24 (KTC 95% từ 0,02 đến 0,45; p=0,025). 65 bệnh nhân (70%) trong nhóm điều trị bằng bosentan có ít nhất 1 tác dụng phụ so với 60 bệnh nhân (65%) trong nhóm giả dược. Nhìn chung, số lượng tác dụng phụ không khác biệt giữa 2 nhóm. Tác dụng phụ thường gặp nhất trong nhóm bosentan là viêm mũi hầu và tăng men gan. Tăng men gan trên 3 lần giới hạn trên của mức bình thường chiếm 13% (12 bệnh nhân) trong nhóm bosentan so với 2% nhóm giả dược. Trong số này, 10 bệnh nhân có tăng men gan trong 20 tuần đầu tiên của điều trị và trở về bình thường khi ngưng hoặc giảm liều bosentan. Để đánh giá tính an toàn của bosentan, 157 trong số 185 bệnh nhân trên được tiếp tục tham gia vào 1 nghiên cứu mở nhãn. Tất cả bệnh nhân được dùng bosentan trong khoảng thời giantrung bình 19,5 tháng (0,1-37,9 tháng).Sau thời gian điều trị chỉ có 7 bệnh nhân có tác dụng phụ nặng có thể liên quan đến bosentan: 1 bn bị suy tim và thiếu máu; 1 bn bị PAH tiến triển; 3 bệnh nhân có tăng men gan và men gan trở về bình thường trên cả 3 bệnh nhân này khi ngưng bosentan. Từ kết quả EARLY cho thấy rằng ngay cả bệnh nhân PAH có triệu chứng nhẹ (WHO-FC II), nếu không được điều trị bệnh có thể diễn tiến xấu về lâm sàng cũng như huyết động, mặc dầu khả năng gắng sức còn bảo tồn và bosentan làm chậm quá trình diễn tiến xấu này. Do đó, bosentan được châu Âu (2008), Canada và Mỹ (2009) chấp thuận cho điều trị bệnh nhân PAH,≥12 tuổivà có WHO-FC II.
Bảng 3: Đặc điểm lâm sàng của bệnh nhân lúc bắt đầu vào nghiên cứu
|
|
Bosentan (n=93) |
Giả dược (n=92) |
|
Giới tính (nam/nữ) |
22 (24%)/71 (76%) |
34 (37%)/58 (63%) |
|
Tuổi (năm) |
45,2 (17,9) |
44,2 (16,5) |
|
Cân nặng (kg) |
67,2 (16,3) |
69 (15,7) |
|
Chủng tộc (da trắng/đen/châu Á) |
87 (94%)/2 (2%)/4 (4%) |
81 (88%)/3 (3%)/8 (9%) |
|
Thời gian từ khi được chẩn đoán PAH đến khi tham gia nghiên cứu này |
2,9 (5,5) |
3,7 (6,5) |
|
Nguyên nhân PAH vô căn |
54 (58%) |
58 (63%) |
|
PAH liên quan với: Tim bẩm sinh |
16 (17%) |
16 (17%) |
|
Bệnh mô liên kết |
18 (19%) |
15 (16%) |
|
Xơ cứng bì |
9 (10%) |
6 (7%) |
|
Lupus |
7 (8%) |
4 (4%) |
|
Sarcoidosis |
1 (1%) |
0 |
|
Bệnh mô liên kết hỗn hợp |
0 |
3 (3%) |
|
Hội chứng Sjogren’s |
0 |
1 (1%) |
|
Viêm động mạch Takayasu’s |
0 |
1 (1%) |
|
Nguyên nhân khác |
1 (1%) |
0 |
|
Nhiễm HIV |
5 (5%) |
2 (2%) |
|
Liên quan với bệnh khác |
0 |
1 (1%) |
|
Điều trị kèm theo Thuốc chống đông máu uống |
55 (59%) |
60 (65%) |
|
Ức chế kênh Ca |
29 (31%) |
38 (41%) |
|
Sildenafil |
14 (15%) |
15 (16%) |
|
6MWD (m) |
438 (86) |
431 (91) |
|
PVR (dyne.s/cm5) |
839 (531) |
805 (369) |
|
Áp lực nhĩ phải (mmHg) |
6,9 (4,5) |
7,5 (5,1) |
|
Áp lực động mạch phổi (mmHg) |
52,5 (18,9) |
52,3 (16) |
|
Chỉ số tim (L/phút/m2) |
2,7 (0,8) |
2,7 (0,6) |
|
Độ bão hòa oxy tĩnh mạch trộn(SVO2) |
66,3 (8,5) |
68 (7,7) |
|
Số liệu được trình bày dưới dạng số lượng n (%) hoặc trung bình (SD: độ lệch chuẩn) |
||
Hình 6: Phần trăm của kháng lực mạch máu phổi sau 6 tháng dùng bosentan và giả dược so với lúc mới vào nghiên cứu
Hình 7: Tỉ lệ bệnh nhân không xuất hiện dấu hiệu trở nặng trên lâm sàng
Hiệu quả bosentan trên tỉ lệ sống còn
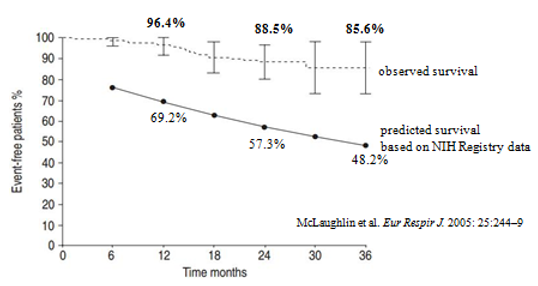
Để đánh giá hiệu quả của bosentan trên tỉ lệ sống còn, 169 bệnh nhân PAH vô cănngay sau khi hoàn tất trong 2 thử nghiệm lâm sàng phân nhóm ngẫu nhiên có đối chứng giả dược 49, 50được McLaughlin và cs tuyển vào một nghiên cứu mở nhãn dùng bosentan đơn trị liệuvà khi cần thiết có thể dùng phối hợp thêm thuốc khác, thời gian điều trị kéo dài 36 tháng53. Tỉ lệ đơn trị bằng bosentan sau 1 và 2 năm điều trị, là 85% và 70%. Tỉ lệ sống còn sau 1 năm và 2 năm điều trị bằng bosentan tương ứng là 96,4% và 88,5% so với tỉ lệ ước đoán là 69% và 57% (hình 8).Cho đến nay, vẫn chưa có nghiên cứu có đối chứng với giả dược về hiệu quả của bosentan trên tỉ lệ sống còn.
Hình 8: Tỉ lệ sống còn trong nhóm dùng bosentan
Hiệu quả và an toàn của bosentan trên bệnh nhi
Thực hiện thử nghiệm lâm sàng có đối chứng trên bệnh nhi PAH gặp rất nhiều khó khăn do: số lượng bệnh nhân ít, không đồng nhất vì là lứa tuổi đang phát triển cả thể chất lẫn tâm thần, các test đánh giá bệnh (như 6MWD, WHO-FC, đánh giá chức năng tim phổi,..) khó thực hiện và có độ tin cậy không cao so với ở người lớn. Do đó chứng cứ hiệu quả của các nhóm thuốc nói chung và bosentan nói riêng trên nhóm dân số này còn hạn chế, chủ yếu đến từ những nghiên cứu mở nhãn. Tuy nhiên từ kết quả của 2 nghiên cứu tiền cứu, đa trung tâm và mở nhãn BREATHE-3 và FUTURE-1, EMA chấp thuận bosentan (dưới dạng tan trong nước) cho điều trị PAHở trẻ em >2 tuổi vào năm 2009. BREATHE-3 đánh giá dung nạp và an toàn sau 12 tuần điều trị bằng bosentan trên 19 bệnh nhi (<15 tuổi) bị PAH, một số bệnh nhân dùng đồng thời eporostenoltruyền TM 54. Các chỉ số về dược lực học và huyết động học được đánh giá trên tất cả bệnh nhi. Kết quả cho thấy bosentan cải thiện các chỉ số huyết động quan trọng và mức tiêu thụ Oxy tối đa (PVO2) cũng như dung nạp tốt (thay đổi mPAP là -8 mmHg; PVR là -300dyne.s/cm5; cả 2 p<0,05).FUTURE-1 đánh giá tính an toàn và dược lực học của thuốc trên 36 bệnh nhi từ 2-12 tuổi bịPAH vô căn hoặc PAH liên quan di truyền55. Kết quả cho thấy bosentan được dung nạp tốt và liều 2mg/kg x 2/ngày có tỉ lệ lợi ích/nguy cơ cao hơn so với liều cao 4 mg x 2/ngày. Về ảnh hưởng bosentan lên sống còn, kết quả từ 1 nghiên cứu hồi cứu tại Anh thực hiện trên 216 bệnh nhi bịPAH vô căn hoặc PAH do nguyên nhân khác cho thấy tỉ lệ sống còn ở nhóm dùng bosentan đơn trị liệu tương đương với nhóm dùng epoprostenol đơn trị56. Thêm vào đó nhiều nghiên cứu khác cũng cho thấy điều trị bosentan lâu dài làm cải thiện chức năng và huyết động cũng như an toàntrên bệnh nhi PAH 57-60.
Liều dùng và cách dùng
Từ những nghiên cứu lâm sàng trên, liều bosentan được khuyến cáo ở người lớn có cân nặng ≥ 40kg liều khởi đầu 62,5mg x 2/ngày trong 4 tuần, sau đó tăng lên liều đích là 125mg x 2/ngày và duy trì lâu dài. Đối với bệnh nhân ≥12 tuổi và <40kg được khuyến cáo dùng liều khởi đầu và liều đích như nhau là 62,5 mgx 2/ngày. Đối với trẻ em, liều khởi đầu 1 mg/kg x 2/ngày sau đó tăng lên liều đích 2 mg x 2/ngày hoặc liều đích 31,25 mg x 2/ngày đối với trẻ có cân nặng 10- 20 kg, 62,5 mg x 2/ngày đối với trẻ 20-40 kg, 125 mg x 2/ngày đối với trẻ > 40 kg, 1/2 liều đích được dùng trong 4 tuần khởi đầu điều trị. Hiện tại nhiều trung tâm điều trị PAH ở Nhật chỉ dùng liều khởi đầu trong 1 tuần, sau đó nếu không thấy tác dụng phụ, sẽ tăng lên liều đích. Men gan được khuyến cáo kiểm tra trước khi bắt đầu điều trị, sau điều trị 1 tháng và lặp lại mỗi tháng.Tránh sử dụng bosentan trên những bệnh nhân có men gan tăng hơn 3 lần mức trên giới hạn bình thường. Nồng độ hemoglobin và hematocrit nên được kiểm tra sau 1 tháng điều trị và theosau mỗi 3 tháng. Đối với phụ nữ trong lứa tuổi sinh nở phải được làm test thử thai trước khi dùng và mỗi tháng trong quá trình điều trị đồng thời phải dùng 2 biện pháp ngừa thai khác nhau ngoại trừ triệt sản và đặt dụng cụ tránh thai.
3. Macitentan
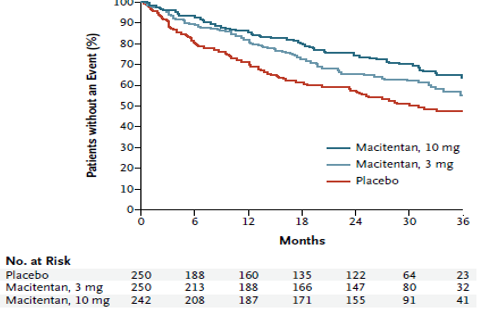
Macitentan là một ERA không chọn lọc mới, được phát triển từ bosentan, làm tăng khả năng gắn kết với thụ thể và xâm nhập mô dẫn đến tăng hiệu quả và giảm tác dụng phụ. Macitentanvừa được FDA và EMA chấp thuận cho điều trị PAH cuối năm 2013 dựa trên kết quả nghiên cứu SERAPHIN. Đây là một thử nghiệm lâm sàng phân nhóm ngẫu nhiên, mù đôi, đa trung tâm và có đối chứng đầu tiên và duy nhất cho đến nay đạt được tiêu chí chính là tiêu chí lâm sàng cứng (tỉ lệ tử vong và bệnh tật): khoảng thời gian từ lúc bắt đầu điều trị đến khi xuất hiện đầu tiên của phối hợp các biến cố chết, cắt vách liên nhĩ, ghép phổi, bắt đầu dùng prostanoids truyền TM hay dưới da, PAH trở nặng. 742 bệnh nhân có tuổi ≥ 12 và WHO-FC từ II đến IV được phân ngẫu nhiên cho dùng giả dược hoặc macitentan 3 mg/ngày hoặc 10 mg/ngày trong 3 năm. Kết quả cho thấy các biến cố thuộc tiêu chí chính xảy ra trên 46,4%, 38% và 31,4% số bệnh nhân tương ứng trong nhóm dùng giả dược, macitentan 3 mg/ngày và 10 mg/ngày (Hình 9). HR của nhóm dùng macitentan 3 mg/ngày so với nhóm giả dược là 0,7 (KTC 97,5% từ 0,52 đến 0,96; p=0.01), đặc biệt HR của nhóm dùng macitentan 10 mg/ngày so với nhóm giả dược là 0,55 (KTC 97,5% từ 0,39 đến 0,76; p<0,001). Điều này có nghĩa macitentan 10 mg/ngày làm giảm 45% nguy cơ kết hợp của tử vong và bệnh tật so với giả dược sau 3 năm điều trị. PAH trở nặng là biến cố thuộc tiêu chí chính thường gặp nhất.Hơn nữa phân tích thêm cho thấy hiệu quả tác dụng của macitentan trêngiảm nguy cơ tử vong và bệnh tậtlà ổn định bất kể khác nhau về giới tính, tuổi, chủng tộc, điều kiện địa lý, bệnh căn, đơn trị liệu hay đa trị liệu phối hợp với những thuốc đặc trị PAH khác và WHO FC (I/II và III/IV). Đối với tiêu chí phụ, macitentan làm giảm 50% nguy cơ tử vong và nhập viện liên quan đến PAH so với giả dược. Tác dụng phụ thường gặp trong nhóm dùng macitentan so với giả dược là nhức đầu, viêm mũi hầu và thiếu máu.
Hình 9: Hiệu quả của macitentan trên tiêu chí chính phối hợp của những biến cố đầu tiên liên quan đến PAH hoặc tử vong do mọi nguyên nhân.
Kết luận
PAH là một bệnh nặng tiến triển gây nên bởi co mạch và tái cấu trúc mạch máu phổi đưa đến dầy thất phải, suy thất phải và tử vong.Bệnh có tỉ lệ tử vong cao và hiện tại chưa có thuốc chữa khỏi. Thuốc điều trị đặc hiệu tác động thông qua 3 con đường cơ chế chính, chủ yếu tác dụng dãn mạch phổi và tác động một phần lên tái cấu trúc mạch máu (chỉ ức chế tăng sinh tế bào cơ trơn mạch máu, không tác động lên tăng sinh tế bào nội mạc). Thuốc làm tăng chất lượng cuộc sống và sống còn từ những nghiên cứu sổ bộ. ERAs đóng một vai trò quan trọng trong điều trị bệnh. Bosentan là một ERA không chọn lọc (ERA), được FDA chấp thuận cho điều trị PAH ở người lớn từ 2001. Thuốc làm cải thiện triệu chứng, khả năng gắng sức, huyết động, kéo dài thời gian dẫn đến diễn tiến xấu trên lâm sàng và có thể tăng tỉ lệ sống còn. Đặc biệt bosentan là thuốc duy nhất cho đến nay dùng trong nghiên cứu (EARLY) là thiết kế chỉ ra lợi ích rất lớn của việc điều trị sớm trên những bệnh nhân có WHOFC II. Bosentan là thuốc có nhiều chứng cứ ủng hộ nhất cho điều trị PAH liên quan bệnh tim bẩm sinh so với những thuốc khác. Thuốc làm cải thiện PVR, mPAP, khả năng gắng sức và không làm thay đổi đáng kể SpO2 trên những bệnh nhân PAH (≥ 12 tuổi) liên quan timbẩm sinh có luồng thông chủ- phổi hoặc hội chứng Eisenmenger. Bosentan cũng được EMA chấp thuận cho điều trị PAH ở trẻ em (≥ 2 tuổi) dựa trên những nghiên cứu cho thấy thuốc làm cải thiện có ý nghĩa mPAP, PVR và mức tiêu thụ tối đa Oxy (PVO2) và dung nạp tốt. Thuốc được dùng nhiều nhất trong điều trị PAH tại Việt Nam hiện nay là sildenafil. Tuy nhiên, sidenafil, chứ không phải bosentan, được FDA khuyến cáo không nên dùng lâu dài trên bệnh nhi PAH từ 1 đến 17 tuổi từ 2012 do tăng tỉ lệ tử vong liên quan đến PAH từ kết quả dài hạn của nghiên cứu START-2 (sử dụng ngắn hạn sildenafil tùy thuộc vào cân nhắc của từng bác sĩ giữa lợi ích và nguy cơ trên bệnh nhân)61-63. Macitentan là một ERA mới không chọn lọc được phát triển từ bosentan, là thuốc đầu tiên và duy nhất cho đến nay làm giảm tỉ lệ tử vong và bệnh tật theo nghiên cứu SERAPHIN (Selexipag, một đồng vận thụ thể prostacyclin cũng làm giảm tỉ lệ tử vong và bệnh tật 39% so với giả dược từ kết quả nghiên cứu GRIPHON, tuy nhiên kết quả đang chờ công bố chính thức). Macitentan làm giảm 45% nguy cơ kết hợp của tử vong và bệnh tật sau 3 năm điều trị.
TÀI LIỆU THAM KHẢO
1. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, Wilkins MR, Badesch DB. Definitions and diagnosis of pulmonary hypertension. Journal of the American College of Cardiology. 2013;62:D42-D50
2. Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. European Respiratory Journal. 2009;34:888-894
3. Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, Gaine SP, Gladwin MT, Jing Z-C, Krowka MJ, Langleben D, Nakanishi N, Souza R. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2009;54:S43-S54
4. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. Journal of the American College of Cardiology. 2013;62:D34-D41
5. Bayer healthcare. Us fda approves bayer’s adempas (riociguat), the first soluble guanylate cyclase stimulator, in two forms of pulmonary hypertension [press release]. October 9, 2013.
6. Fang JC, DeMarco T, Givertz MM, Borlaug BA, Lewis GD, Rame JE, Gomberg-Maitland M, Murali S, Frantz RP, McGlothlin D, Horn EM, Benza RL. World health organization pulmonary hypertension group 2: Pulmonary hypertension due to left heart disease in the adult—a summary statement from the pulmonary hypertension council of the international society for heart and lung transplantation. The Journal of Heart and Lung Transplantation.31:913-933
7. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. New England Journal of Medicine. 2004;351:1425-1436
8. Tuder RM, Yeager ME, Geraci M, Golpon HA, Voelkel NF. Severe pulmonary hypertension after the discovery of the familial primary pulmonary hypertension gene. European Respiratory Journal. 2001;17:1065-1069
9. Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. European Respiratory Journal. 2012;40:1555-1565
10. Rabinovitch M. Pathobiology of pulmonary hypertension. Annual Review of Pathology: Mechanisms of Disease. 2007;2:369-399
11. Hung TV, Emoto N, Vignon-Zellweger N, Nakayama K, Yagi K, Suzuki Y, Hirata K-i. Inhibition of vascular endothelial growth factor receptor under hypoxia causes severe, human-like pulmonary arterial hypertension in mice: Potential roles of interleukin-6 and endothelin. Life Sciences.
12. Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, Reid LM, Tuder RM. Pathologic assessment of vasculopathies in pulmonary hypertension. Journal of the American College of Cardiology. 2004;43:S25-S32
13. Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. The American Journal of Pathology. 1994;144:275-285
14. Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982–2006. European Respiratory Journal. 2007;30:1103-1110
15. Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the reveal registry. Chest. 2012;142:448-456
16. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411-415
17. Inoue A, Yanagisawa M, Kimura S, Kasuya Y, Miyauchi T, Goto K, Masaki T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2863-2867
18. Mattoli S, Mezzetti M, Riva G, Allegra L, Fasoli A. Specific binding of endothelin on human bronchial smooth muscle cells in culture and secretion of endothelin-like material from bronchial epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 1990;3:145-151
19. Yu JC, Davenport AP. Secretion of endothelin-1 and endothelin-3 by human cultured vascular smooth muscle cells. British Journal of Pharmacology. 1995;114:551-557
20. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. The Journal of Experimental Medicine. 1990;172:1741-1748
21. Miyauchi T, Masaki T. Pathophysiology of endothelin in the cardiovascular system. Annual Review of Physiology. 1999;61:391-415
22. Masaki T. The discovery of endothelins. 1998.
23. Shinmi O, Kimura S, Sawamura T, Sugita Y, Yoshizawa T, Uchiyama Y, Yanagisawa M, Goto K, Masaki T, Kanazawa I. Endothelin-3 is a novel neuropeptide: Isolation and sequence determination of endothelin-1 and endothelin-3 in porcine brain. Biochemical and Biophysical Research Communications. 1989;164:587-593
24. Levin ER. Endothelins. New England Journal of Medicine. 1995;333:356-363
25. Clozel M, Clozel JP. Effects of endothelin on regional blood flows in squirrel monkeys. Journal of Pharmacology and Experimental Therapeutics. 1989;250:1125-1131
26. Archer S, Rich S. Primary pulmonary hypertension: A vascular biology and translational research “work in progress”. Circulation. 2000;102:2781 – 2791
27. Kedzierski RM, Yanagisawa M. Endothelin system: The double-edged sword in health and disease. Annual Review of Pharmacology and Toxicology. 2001;41:851-876
28. Lüscher TF, Barton M. Endothelins and endothelin receptor antagonists: Therapeutic considerations for a novel class of cardiovascular drugs. Circulation. 2000;102:2434-2440
29. Seo B, Oemar BS, Siebenmann R, von Segesser L, Lüscher TF. Both eta and etb receptors mediate contraction to endothelin-1 in human blood vessels. Circulation. 1994;89:1203-1208
30. Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. The FASEB Journal. 1995;9:1196-1204
31. Ohlstein EH, Arleth A, Bryan H, Elliott JD, Cheng Po S. The selective endothelin eta receptor antagonist bq123 antagonizes endothelin-1-mediated mitogenesis. European Journal of Pharmacology: Molecular Pharmacology. 1992;225:347-350
32. Sugawara F, Ninomiya H, Okamoto Y, Miwa S, Mazda O, Katsura Y, Masaki T. Endothelin-1-induced mitogenic responses of chinese hamster ovary cells expressing human endothelina: The role of a wortmannin-sensitive signaling pathway. Molecular Pharmacology. 1996;49:447-457
33. Hirata Y, Emori T, Eguchi S, Kanno K, Imai T, Ohta K, Marumo F. Endothelin receptor subtype b mediates synthesis of nitric oxide by cultured bovine endothelial cells. Journal of Clinical Investigation. 1993;91:1367-1373
34. Naomi S, Iwaoka T, Disashi T, Inoue J, Kanesaka Y, Tokunaga H, Tomita K. Endothelin-1 inhibits endothelin-converting enzyme-1 expression in cultured rat pulmonary endothelial cells. Circulation. 1998;97:234-236
35. Dupuis J, Stewart DJ, Cernacek P, Gosselin G. Human pulmonary circulation is an important site for both clearance and production of endothelin-1. Circulation. 1996;94:1578-1584
36. Dupuis J, Jasmin JF, Prié S, Cernacek P. Importance of local production of endothelin-1 and of the etbreceptor in the regulation of pulmonary vascular tone. Pulmonary Pharmacology & Therapeutics. 2000;13:135-140
37. Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H, Schäfers H-J. Selective upregulation of endothelin b receptor gene expression in severe pulmonary hypertension. Circulation. 2002;105:1034-1036
38. Benza RL, Mehta S, Keogh A, Lawrence EC, Oudiz RJ, Barst RJ. Sitaxsentan treatment for patients with pulmonary arterial hypertension discontinuing bosentan. The Journal of Heart and Lung Transplantation. 2007;26:63-69
39. Opitz CF, Ewert R, Kirch W, Pittrow D. Inhibition of endothelin receptors in the treatment of pulmonary arterial hypertension: Does selectivity matter? European Heart Journal. 2008;29:1936-1948
40. Rubens C, Ewert R, Halank M, Wensel R, Orzechowski H-D, Schultheiss H-P, Hoeffken G. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension*. Chest. 2001;120:1562-1569
41. Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Annals of Internal Medicine. 1991;114:464-469
42. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, Kimura S, Masaki T, Duguid WP, Stewart DJ. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. New England Journal of Medicine. 1993;328:1732-1739
43. Yoshibayashi M, Nishioka K, Nakao K, Saito Y, Matsumura M, Ueda T, Temma S, Shirakami G, Imura H, Mikawa H. Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Evidence for increased production of endothelin in pulmonary circulation. Circulation. 1991;84:2280-2285
44. Ishikawa S, Miyauchi T, Sakai S, Ushinohama H, Sagawa K, Fusazaki N, Kado H, Sunagawa H, Honda S, Ueno H, Yamaguchi I, Sugishita Y, Goto K. Elevated levels of plasma endothelin-1 in young patients with pulmonary hypertension caused by congenital heart disease are decreased after successful surgical repair. The Journal of Thoracic and Cardiovascular Surgery.110:271-273
45. Lutz J, Gorenflo M, Habighorst M, Vogel M, Lange Peter E, Hocher B. Endothelin-1- and endothelin-receptors in lung biopsies of patients with pulmonary hypertension due to congenital heart disease. Clinical Chemistry and Laboratory Medicine. 1999;37:423
46. Sitbon O, Humbert M, al. e. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: Prognostic factors and survival. J Am Coll Cardiol. 2002;40(4):780-788
47. Kane G, Maradit-Kremers H, al. e. Integration of clinical and hemodynamic parameters in the prediction of long-term survival in patients with pulmonary arterial hypertension. Chest. 2011;139(6):1285-1293
48. Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, McGoon MD, McLaughlin VV, Roecker EB, Gerber MJ, Dufton C, Wiens BL, Rubin LJ, for the Ambrisentan in Pulmonary Arterial Hypertension R, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies Group. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (aries) study 1 and 2. Circulation. 2008;117:3010-3019
49. Channick RN, Simonneau G, Sitbon O, Robbins IM, Frost A, Tapson VF, Badesch DB, Roux S, Rainisio M, Bodin F, Rubin LJ. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: A randomised placebocontrolled study. The Lancet. 2001;358:1119-1123
50. Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. New England Journal of Medicine. 2002;346:896-903
51. Galiè N, Beghetti M, Gatzoulis MA, Granton J, Berger RMF, Lauer A, Chiossi E, Landzberg M, Investigators ftBRToEAT-. Bosentan therapy in patients with eisenmenger syndrome: A multicenter, double-blind, randomized, placebo-controlled study. Circulation. 2006;114:48-54
52. Galiè N, Rubin LJ, Hoeper MM, Jansa P, Al-Hiti H, Meyer GMB, Chiossi E, Kusic-Pajic A, Simonneau G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (early study): A double-blind, randomised controlled trial. The Lancet.371:2093-2100
53. McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galiè N, Rainisio M, Simonneau G, Rubin LJ. Survival with first-line bosentan in patients with primary pulmonary hypertension. European Respiratory Journal. 2005;25:244-249
54. Barst R, Ivy D, Dingemanse J, Widlitz A, et a. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther. 2003;Apr;73(4):372-82.
55. Beghetti M, Haworth SG, Bonnet D, Barst RJ, Acar P, Fraisse A, Ivy DD, Jais X, Schulze-Neick I, Galiè N, Morganti A, Dingemanse J, Kusic-Pajic A, Berger RMF. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: The future-1 study. British Journal of Clinical Pharmacology. 2009;68:948-955
56. Haworth SG, Hislop AA. Treatment and survival in children with pulmonary arterial hypertension: The uk pulmonary hypertension service for children 2001–2006. Heart. 2009;95:312-317
57. Rosenzweig EB, Ivy DD, Widlitz A, Doran A, Claussen LR, Yung D, Abman SH, Morganti A, Nguyen N, Barst RJ. Effects of long-term bosentan in children with pulmonary arterial hypertension. Journal of the American College of Cardiology. 2005;46:697-704
58. Maiya S, Hislop AA, Flynn Y, Haworth SG. Response to bosentan in children with pulmonary hypertension. Heart. 2006;92:664-670
59. Ivy DD, Rosenzweig EB, Lemarié J-C, Brand M, Rosenberg D, Barst RJ. Long-term outcomes in children with pulmonary arterial hypertension treated with bosentan in real world clinical settings. The American journal of cardiology. 2010;106:1332-1338
60. Hislop AA, Moledina S, Foster H, Schulze-Neick I, Haworth SG. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. European Respiratory Journal. 2011;38:70-77
61. Barst RJ, Beghetti M, Pulido T, Layton G, Konourina I, Zhang M, Ivy DD. Starts-2: Long-term survival with oral sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation. 2014;129:1914-1923
62. Fda drug safety communication: Fda recommends against use of revatio (sildenafil) in children with pulmonary hypertension. FDA 2012
63. Fda drug safety communication: Fda clarifies warning about pediatric use of revatio (sildenafil) for pulmonary arterial hypertension. FDA 2014


{kind=link}