

 Bệnh cao huyết áp ngày càng gia tăng trên thế giới. Theo thống kê của Hiệp hội Tim mạch Hoa Kỳ năm 2006 thì có đến 73,5 triệu người trên 20 tuổi mắc bệnh cao huyết áp: cứ 3 người trưởng thành tại Hoa kỳ thì có 1 người bị cao huyết áp.
Bệnh cao huyết áp ngày càng gia tăng trên thế giới. Theo thống kê của Hiệp hội Tim mạch Hoa Kỳ năm 2006 thì có đến 73,5 triệu người trên 20 tuổi mắc bệnh cao huyết áp: cứ 3 người trưởng thành tại Hoa kỳ thì có 1 người bị cao huyết áp.
Viện tim TP.HCM
I. TỔNG QUAN
Bệnh cao huyết áp ngày càng gia tăng trên thế giới. Theo thống kê của Hiệp hội Tim mạch Hoa Kỳ năm 2006 thì có đến 73,5 triệu người trên 20 tuổi mắc bệnh cao huyết áp: cứ 3 người trưởng thành tại Hoa kỳ thì có 1 người bị cao huyết áp. Trong vòng 10 năm, từ 1996 đến 2006 tỷ lệ cao huyết áp tại Hoa Kỳ tăng gần 20%, và tỷ lệ tử vong do cao huyết áp tăng 48%[ 1]. Tại Việt Nam, tại ” Hội nghị sơ kết sơ kết dự án phòng, chống tăng huyết áp năm 2009 và kế hoạch triển khai 2010″ do Bộ Y tế Việt Nam tổ chức vào tháng 4 năm 2009 tại Hà Nội thì : theo khảo sát ngẫu nhiên đối với người dân từ 25 tuổi trở lên ở Hà Nội, Thái Nguyên, Thái Bình và Nghệ An, tần suất tăng huyết áp đã tăng đến 16,3%; trong đó tỷ lệ tăng huyết áp ở thành thị là 22,7% và ở nông thôn là 12,3% [2,3]. Cao huyết áp là một trong những yếu tố nguy cơ dễ dẫn đến hình thành mảng xơ vữa và tiến triển bệnh lý cơ tim thiếu máu cục bộ, NMCT và tai biến mạch máo não: tại Hoa Kỳ huyết áp >140/90mmHg thấy được trên 69% số bệnh nhân bị NMCT lần đầu, 77% trên những bệnh nhân bị tai biến mạch máu não lần đầu và 74% những bệnh nhân bị suy tim. Nguyên nhân chính trong cao huyết áp dẫn đến hình thành mãng xơ vữa động mạch là sự rối loạn chức năng nội mạc, mất cân bằng giữa quá trình chết và tái tạo tế bào nội mạc mạch máu [4,5].
II. NỘI MẠC MẠCH MÁU:
Nội mạc mạch máu là một lớp tế bào nằm bên trong lòng mạch máu: ngăn cách giữa các tế bào máu lưu thông trong lòng mạch và các tế bào cơ trơn của mạch máu. Bình thường trong cơ thể một người trưởng thành tất cả lớp nội mạc mạch máu cân nặng khoảng 1.5kg và có diện tích khoảng 800-100m2. Nội mạc máu đóng vai trò quan trọng trong điều hòa sự co và dãn mạch, do đó có thể kiểm soát áp lực máu trong lòng mạch; đóng vai trò trong cân bằng sự hình thành huyết khối và tiêu huyết khối, giúp ngăn chặn tạo huyết khối trong lòng mạch, tạo sự cân bằng chống phản ứng viêm trong lòng mạch máu, chống tăng sinh cơ trên dưới lớp nội mạc[6]. Các tế bào nội mạc mạch vành có khả năng truyền tải các tín hiệu hóa sinh từ máu, cảm nhận các lực cơ học trong lòng mạch và điều chỉnh sự co hoặc dãn mạch máu thông qua sự sản xuất một loạt các yếu tố dịch thể tác động lên mạch máu làm dãn mạch như ” yếu tố làm dãn mạch nội mạc” (EDRF), nitric oxide (NO), prostacylin …, chất làm cho mạch như endothelin 1 (ET-1) , angiotensin II,… Đối với mạch vành: khi vận động mạch vành có thể dãn từ 2 đến 5 lần làm tăng lượng máu đến cơ tim, nhưng khi mạch vành bị mảng xơ vữa gây hẹp lòng mạch vành làm mất cân bằng giữa sự sản xuất chất dãn mạch và chất gây co mạch máu. Khi mạch vành hẹp 70% thì khả năng dãn mạch trong lúc vận động không còn làm người bệnh đau ngực khi vận động là cơn đau tức ngực ổn định. Nếu mảng xơ vữa bị bong ra tạo huyết chối gây tắc mạch vành không có tưới máu sau chỗ tắc thì bệnh nhân bị Hội chứng vành cấp (NMCT ST chênh lên, NMCT không ST chênh lên, cơn đau thắt ngực không ổn định).
Bình thường thì Nitric oxide (NO), prostacyclin của nội mạc mạch máu làm dãn mạch, có tính kháng viêm, chống tăng sinh tế bào, và ức chế sự hình thành huyết khối (cùng với t-PA, TFPI tiết ra từ tế bào nội mạc). Dưới ảnh hưởng của các yếu tố nguy cơ như cao huyết áp gây giảm sản xuất NO, tăng tiết endothelin gây co mạch, tăng phản ứng viêm, làm tăng sinh tế bào và tăng nguy cơ hình thành huyết khối (cùng với sự giảm tiết t-PA, tăng tiết PAI). Rối loạn chức năng nội mạc có thể phát hiện được bằng những thay đổi trong đáp ứng co dãn mạch máu với các chất vận mạch, sự tăng sinh tế bào, sự kết tập tiểu cầu, tính thấm thành mạch hoặc sự tương tác giữa bạch cầu và tế bào nội mạc [7]. Trong các nghiên cứu khoa học và thử nghiệm lâm sàng, đánh giá rối loạn chức năng nội mạc là đánh giá chức năng co hoặc dãn mạch, đặc biệt là chức năng dãn mạch phụ thuộc vào nội mạc. Trong lâm sàng, đánh giá rối loạn chức năng nội mạc bằng đo nồng độ các chất chỉ thị của nội trong máu như NO, endothelin-1, các chất chỉ thị phản ứng viêm như “phân tử kết dính nội tế bào”, seleclin… Các nghiên cứu gần đây về sinh học tế bào sử dụng các chất chỉ thị trong máu như các mảnh nhỏ tế bào nội mạc chết theo chương trình và các tiền chết tạo ra tế bào nội mạc, cũng như chính bản thân các tế bào nội mạc trong máu.
III. CAO HUYẾT ÁP VÀ HÌNH THÀNH MẢNG XƠ VỮA ĐỘNG MẠCH:
Rất nhiều nghiên cứu cho thấy giảm sản xuất NO và tăng quá trình chết theo chương trình của tế bào nội mạc (Apoptosis) là một trong những nguyên nhân chính gây loạn chức năng nội mạc dẫn đến hình thành mãng xơ vữa động mạch. Trên những bệnh nhân cao huyết áp thì giảm NO do giảm sản xuất NO, và tăng quá trình ức chế NO đã được tiết ra. Giảm sản xuất NO do: (1) Giảm L-arginin là tiền chất tổng hợp nên NO synthase [8,9], (2) Sản xuất nhiều trong tế bào ADMA (Asymetric dimethylArginine) là chất ức chế cạnh tranh NO synthase, dẫn đến giảm sản xuất NO [10,11], (3) Thiếu cộng tố (cofactor ) để tổng hợp eNOS (endothelial NO synthase). Giảm hoạt tính của NO đã được tiết ra chủ yếu do tế bào dưới ảnh hưởng của cao huyết áp sản sinh nhiều Superoxide O 2- [12,13].
Các tế bào nội mạc mạch máu nói chung và mạch vành nói riêng luôn luôn diễn ra quá trình chết tế bào theo chương trình và tân tạo tế bào mới giúp cho lớp nội mạc luôn liền lạc và duy trì các chức năng của lớp nội mạc. Dưới ảnh hưởng của cao huyết áp các tế bào nội mạc chết theo chương trình (apoptosis) nhiều hơn tạo điều kiện cho hình thành mảng xơ vữa động mạch [14]. Cơ chế chính làm quá trình apoptosis của nội mạc mạch chết theo chương trình nhiều hơn là sự tăng tạo angiotensin II nhiều trong máu và trong mô tế bào. Theo tác giả Sealey JE tại Trung tâm Y khoa C
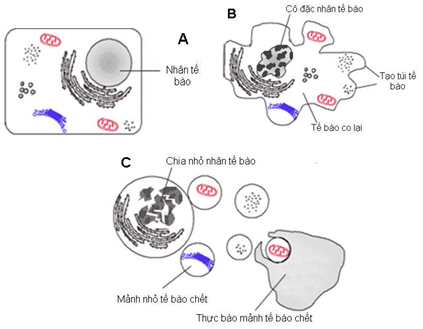
ornell-Newyork- Hoa kỳ thì trên những bệnh nhân cao huyết áp tiên phát các tiểu cầu thận tại thận chia làm 2 nhóm: (1) một nhóm tiểu cầu thận thiếu máu tưới do hẹp tiểu động mạch đến tiểu cầu thận dẫn đến tăng tiết Renin mãn tính (do số lượng nhóm tiểu cầu thận thiếu máu này ít nên đa số bệnh nhân cao huyết áp trong một thời gian dài không có biểu hiện suy chức năng thận). Sự tăng tiết renin của nhóm tiểu cầu thận thiếu máu dẫn đến tăng Aldosterone và tăng tái hấp thụ Natri; (2) Để điều hòa sự tăng tiết renin do nhóm tiểu cầu thận thiếu máu gây nên, các tiểu cầu thận bình thường tăng mức lọc máu qua tiểu cầu thận để tăng thải Natri. Sự tăng bài tiết Natri ở các tiểu cầu thận bình thường gây ra sự ức chế ngược làm ức chết bài tiết renin ở các tiểu cầu thận bình thường này [15,16]. Tuy nhiên khi tăng độ lọc tiểu cầu thận ở các tiểu cầu thận bình thường làm tăng áp suất thủy tĩnh mao mạch tiểu cầu, điều này làm co tiểu động mạch vào tiểu cầu thận một cách tự động để giới hạn dòng máu vào tiểu cầu thận ổ ạt quá mức [17]. Đồng thời khi tăng lọc ở tiểu cầu thận thì các tế bào ống thận gần tăng tiết Angiotensinogen tại chỗ [18,19,20] . Sự co tự động tiểu động mạch vào tiểu cầu thận ở các tiểu cầu thận bình thường và tiết Angiotensinogen tại chỗ ở tế bào ống thận gần khi tăng độ lọc ở tiểu cầu thận gây mất cân bằng cơ chế điều hòa sự tăng tiết renin ở các tiểu cầu thận thiếu máu, dẫn đến tăng nồng độ Angiotensin II trong máu. Angiotensin II làm tăng quá trình Apoptosis của tế bào nội mạc mạch máu thông qua sự kích hoạt CASP 3 là một protein thuộc họ caspase (Cysteine aspartic acid protease) và làm tăng Protein kinase C trong tế bào làm tế bào bắt đầu quá trình apoptosis [21,22] (H1.)
Hình 1: Quá trình Apoptosis của tế bào nội mạc (a) tế bào bình thường, (b) co tế bào và cô đặc nhân, (c) chia nhỏ nhân tế bào và tế bào, sau đó các mảng nhỏ được thực bào.
Tác giả Stefamie Dimmeler (tại khoa Nội tim mạch. Đại học Frankfurt- Đức) nghiên cứu ủ nội mạc của mạch máu cuống rốn với Angiotensin II (nồng độ 1umol/l ) trong 18 giờ, đã nhận thấy Angiotensin II làm tăng tỷ lệ apoptosis của tế bào nội mạc từ 1,7 ± 2% (nhóm chứng) lên 4,5 ±1,7% (nhóm có Angiotensin II) (P<0,05). Trong nghiên cứu: khi tăng nồng độ angiotensin II làm tăng tỷ lệ tế bào nội mạc bị aopotosis, trong khi đó tế bào cơ trơn không bị tăng tỷ lệ apotosis khi tăng nồng độ angiotensin II ( H.2). Tác giả cũng chứng minh được là Angiotensin II kích hoạt CASP 3 làm bắt đầu quá trình apotosis tế bào nội mạc. Khi sử dụng chất ức chế peptide caspase 3 là AC-DEVD-CHO thì Angiotensin II không còn làm tăng quá trình apoptosis của tế bào nội mạc giữa [21].
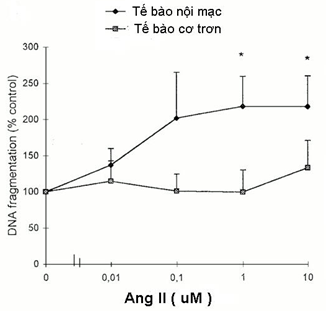
Hình 2: Khi tăng nồng độ angiotensin II thì tăng tỷ lệ tế bào nội mạc bị aopotosis, trong khi đó tế bào cơ trơn không tăng tỷ lệ apotosis khi tăng nồng độ angiotensin II
Tác giả Dayuan Li tại khoa Y- Đại học Flonda -Hoa Kỳ khi ủ tế bào nội mạc mạch vành người với Angiotensin II trong 27 giờ nhận thấy Angiotensin II kích hoạt protein kinase C, protein tyrosin kinase làm bắt đầu quá trình apoptosis của tế bào nội mạc mạch vành. Nếu ủ nội mạc mạch vành người với chất ức chế protein kinase C và Angiotensin II , thì tỷ lệ tế bào bị apoptosis giảm đi so với chỉ ủ với angiotensin II (biểu đồ 1)[22].
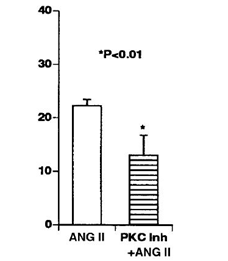
Biểu đồ 1: Tế bào nội mạc mạch vành người ủ với chất ức chế protein kinase C làm giảm tỷ lệ tế bào bị apotosis so với chỉ ủ với angiotensin II
Tác giả Werner tại Đại học Hamburg-Đức nhận thấy, khi tăng nồng độ angiotensin II trong máu tác động lên tiếp nhận thể AT-1, dẫn đến hoạt hóa chuỗi phản ứng NADH/NAD(P)H trong tế bào nội mạc [23]. NAD(P)H oxidase là men liên kết với màng tế bào làm chuyển 1 electron của NADH hoặc NAD(P)H trong tế bào đến phân tử Oxy: NAD(P)H + 2O 2gNAD(P)+ + H+ + 2O–. Khi Superoxide được sản xuất ra quá nhiều, dẫn đến tình trạng quá tải superoxide và gọi là ” Stress oxy hóa” ( stress oxidative). Stress oxy hóa làm thay đổi DNA của nhân tế bào, oxy hóa Lipid và Cholesterol, thay đổi cấu trúc của một số protein, và kích hoạt các tế bào kết dính mạch máu, phân tử kết dính tế bào và phân tử hấp dẫn monocyte ( ICAM-1, VCAM-1, MCP-1), làm tăng phản ứng viêm. Những thay đổi khi ăng Superoxide do cao huyết áp dẫn đến hình thành mảng xơ vữa động mạch [25-28]. Tác giả Makoto Usuii tại Viện nghiên cứu Tim mạch Kyushu-Fukuoka-Nhật, khi nghiên cứu tác động của chất ức chế tổng hợp NO ( L-NAME: N-nitro-L-arginine methyl ester) và chất ức chế tiếp nhận thể AT-1 trên 5 nhóm mèo, mỗi nhóm 5 con: nhóm 1 là nhóm chứng, nhóm 2 được uống chất ức chế tổng hợp NO; nhóm 3 và 4 được uống chất ức chế tổng hợp NO với chất ức chế tiếp nhận thể AT-1 (lượng chất ức chế tiếp nhận thể AT-1 ở nhóm 3 cao hơn nhóm 4), nhóm 5 được cho uống L-NAME và tiêm chất chống oxy hóa là Thiol antioxidant + N acetylcystein vào màng bụng. Sau 3 ngày nghiên cứu, kết quả nghiên cứu tế bào dưới kính hiển vi điện tử cho thấy ở nhóm uống chất ức chế sản xuất NO có thâm nhiễm nhiều tế bào monocyte, MCP-1, IgG , có phản ứng viêm mô kẽ tại lớp nội mạch mạch vành; trong khi đó ở nhóm chứng và các nhóm có uống chất ức chế AT-1, cũng như chích antioxidant thì không có hiện tượng đó (H.3). Diện tích vùng mạch vành bị thâm nhiễm tế bào viêm quan sát thấy nhiều nhất trên nhóm chỉ có uống L-NAME là chất ức chế sản xuất NO (biểu đồ 2).

Hình 3: Mạch vành của nhóm uống chất ức
chế sản xuất NO quan sát thấy có thâm nhiễm nhiều tế bào monocyte, có phản ứng viêm mô kẽ tại lớp nội mạch mạch vành
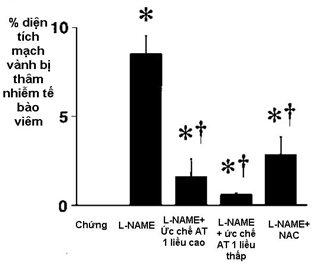
Biểu đồ 2: Diện tích mạch vành bị thâm nhiễm tế bào viêm quan sát thấy nhiều nhất trên nhóm chỉ có uống L-NAME là chất ức chế sản xuất NO.
Nghiên cứu của tác giả Makoto Usuii cho thấy giảm sản xuất NO làm tăng phản ứng viêm, tăng MCP-1 dẫn đến dễ hình thành mảng xơ vữa động mạch tại mạch vành. Khi ức chế tiếp nhận thể AT-1 thì phản ứng viêm không xẩy ra, như vậy nếu giảm sự sản xuất angiotensin II sẽ dẫn đến giảm nguy cơ xơ vữa động mạch. Tác giả Hiroshi Miyazaki tại khoa nội tim mạch-Đại học Y khoa Kurume-Nhật nghiên cứu trên 118 người (tuổi từ 26 đến 77), trong đó có 40 bệnh nhân cao huyết áp. Kết quả cho thấy nồng độ asymmetric dimethylarginine (ADMA) tương quan thuận với chỉ số huyết áp của bệnh nhân (Biểu đồ 3). ADMA là chất ức chế với NO synthase, làm giảm sản xuất NO; như vậy khi huyết áp càng cao thì nội mạc sản xuất càng nhiều ADMA, gây ức chế sản xuất NO [29].

Biểu đồ 3: Nồng độ ADMA tương quan với chỉ số huyết áp.
Những nghiên cứu trên cho thấy khi tăng huyết áp làm tăng nồng độ angiotesin II, giảm sản xuất NO, tăng stress oxy hóa, dẫn đến tăng LDL-oxy hóa. Đồng thời angiotensin II cũng làm tăng quá trình apoptosis, làm màng tế bào nội mạc không liền lạc, dẫn đến phân tử LDL oxy hóa dễ đi vào lớp dưới nội mạc. Angiotensin II tác động lên tiếp nhân thể AT-1 làm tăng stress oxy hóa và tăng phản ứng viêm, kích hoạt các phân tử kết dính như ICAM-1, VCAM-1, phân tử hấp dẫn tế bào monocyte, tạo thành tế bào bọt dưới nội mạc, cuối cùng là hình thành mảng xơ vữa động mạch (hình 4).

Hình 4: (A) Stress oxy hóa và apoptosis bắt đầu quá trình làm rối loạn chức năng lớp nội mạc mạch vành. ( B) hình thành tế bào bọt do macrophage thực bào LDL oxy hóa. ( C ) hình thành mãng xơ vữa, ( D ) Bong tróc mãng xơ vữa do tăng thể tích nhiều trong lõi Lipid, tăng phản ứng viêm, tăng apoptosis.
Đối với chứng bệnh nhân đã hẹp mạch vành thì sự tăng lên đáng kể của quá trình apoptosis đóng góp một phần làm mảng xơ vữa dễ vỡ, tạo huyết khối và gây nên hội chứng vành cấp [30-32]. Nghiên cứu của tác giả Marco Valgimigli tại khoa tim mạch trường Đại học Ferrara- Ý trên 154 người chia làm 3 nhóm: nhóm (1) có 40 người bình thường (tuổi trung bình 65 ± 6; có 23 nam, không có tiền sử bệnh và không được điều trị gì); nhóm (2) có 32 bệnh nhân (tuổi trung bình 68 ± 11) với cơn đau thắt ngực ổn định độ II hoặc III theo phân loại của Hiệp hội tim mạch Canada; những bệnh nhân thuộc nhóm 2 được chụp mạch vành và được chứng minh là có tổn thương mạch vành; nhóm (3) có 41 bệnh nhân (tuổi trung bình 67± 10 ) với hội chứng vành cấp (nhóm 3 lại chia 2 phân nhóm: (3A) gồm 14 bệnh nhân với cơn đau thắt ngực không ổn định và phân nhóm (3B) gồm 24 bệnh nhân NMCT với ST chênh lên và 3 bệnh nhân NMCT không ST chênh lên). Tác giả ủ huyết tương của các người bình thường và bệnh nhân tham gia nghiên cứu với nội mạc tĩnh mạch rốn: kết quả cho thấy ở nhóm bệnh nhân hội chứng vành cấp có tỷ lệ tế bào chết theo chương trình (apoptosis) cao nhất (14 ± 6%) so với nhóm bệnh nhân với cơn đau thắt ngực ổn định (3,31 ± 1,8%) và người bình thường (1,35 ± 0,8%) (P<0,001) ( H.5). Tác giả Marco Valgimigli cũng nhận thấy tỷ lệ tế bào nội mạc bị apoptosis tương quan thuận với hình thái tổn thương mạch vành (r=0,58, P<0,005): những bệnh nhân với 1 tổn thương phức tạp có tỷ lệ tế bào nội mạc bị apoptosis là 10 ± 4,7%, trong khi đó những bệnh nhân với nhiều tổn thương phức tạp có tỷ lệ tế bào nội mạc bị apoptosis là 17,5 ± 4,6% ( P< 0,0001) [33].
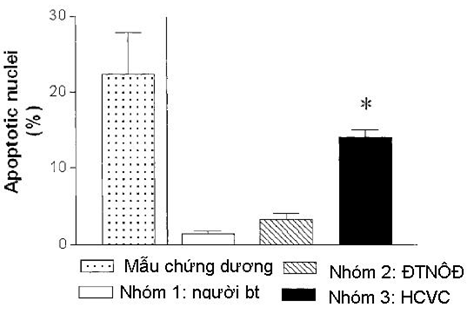
Hình 5: Nhóm BN HCVC( nhóm 3) có tỷ lệ tế bào apoptosis cao nhất so với nhóm BN với cơn đau thắt ngực ổn định ( nhóm 2) và người bình thường (nhóm 1)
IV. KẾT LUẬN:
Cao huyết áp làm giảm sản xuất NO, tăng stress oxy hóa, dẫn đến tăng LDL-oxy hóa. Cao huyết áp còn làm tăng quá trình apoptosis tế bào nội mạc mạch máu do tăng sản xuất angiotensin II làm màng tế bào nội mạc không liền lạc giúp phân tử LDL oxy hóa dễ dàng đi vào lớp dưới nội mạc. Angiotensin II tác động lên tiếp nhân thể AT-1 làm tăng phản ứng viêm, và kích hoạt các phân tử kết dính như ICAM-1, VCAM-1, phân tử hấp dẫn tế bào monocyte, tạo thành tế bào bọt dưới nội mạc, cuối cùng là hình thành mảng xơ vữa động mạch. Trên những bệnh nhân đã bị hẹp mạch máu do xơ vữa động mạch thì quá trình apoptosis do cao huyết áp làm mãng xơ vữa dễ nứt vỡ, tạo huyết khối, gây nên hội chứng vành cấp. Do đó cần thiết điều trị tích cực, kiểm soát tốt huyết áp trên những bệnh nhân bị cao huyết áp, và tích cực tuyên truyền phòng ngừa cao huyết áp trên những người bình thường có nguy cơ dẫn đến cao huyết áp.
TÀI LIỆU THAM KHẢO.
1/Cardiovascular Disease Statistics of American Heart Association 2006. http://grfw.org/.
2/Hội nghị sơ kết sơ kết dự án phòng, chống tăng huyết áp năm 2009 và kế hoạch triển khai 2010. Bộ y tế- Việt Nam
3/Khuyến cáo 2008 về các bệnh lý tim mạch và chuyển hóa. Nhà xuất bản y học – Chi nhánh TP.HCM -2008.
4/Braunwald’s Heart Disease. A text book of Cardovascular Medicine. Seventh edition. Elservier Saunders. 1103-1127.
5/ Claudio Ceconi, Kim M Fox. ACE inhibition with peridopril and endothelial function. Results of a substudy of the Europa study: PERTINENT. Cardionvascular Research 73 (2007): 237 – 46.
6/ Celermaper DS. Endothehal dysfurction: does it matter ? Is it reversible ? JACC 1997; 30: 325 -333
7/ C. Thuillez, V. Richard. Targeting endothelial dysfunction in hypertensive subjects. Journal of Human Hpertension ( 2005) 19, S21- 25.
8/ Zhou MS et al. L-Arginine improves endothelial function in renal artery of hypertensive Dahl rats. J Hypertens 2001; 19: 421-429.
9/Dimitris Tousoulis,Charalambos Antoniades.L-Arginine in cardiovascular disease: dream or reality? Vasc Med 2002 7: 203-207.
10/Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol 2000; 20: 2032-2037.
11/ Surdacki S et al. Reduced urinary excretion of nitric oxide metabolites and increases plasma levels of asym- metric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol 1999; 33: 652-658.
12/Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdown of endothelium derived relaxing factor. Nature 1986; 320: 454-460.
13/Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactive endothelium derived relaxing factor. Am J Physiol 1986; 250: H822-H827.
14/ Braunwal’s heart disease. Atherosclerate cardiovascular disease. seven Edition. Elsevier saunders 2005: 113 – 1139.
15/ Sealey JE, Blumenfeld JD, Bell GM. On the renal basis for essential hypertension : nephoun heterogeneity with discordant renin secretion and sodium excretion causing a hypertensive vasoconstrictstion volume relation ship. J Hyperten. 1988 oct: 6 (10): 763-77
16/ Laragh JH. Discordant nephron function. A pathogemic factor in hypertesion and it’s vascular comphication of stroke and heart attack. Am J Hypertens; 1991 Jan; 4 (1pt2): 2S- 6S.
17/ Guyton Arthur C. Textbook of Medical physiology, 10th ed; Saunders Elsevier, philandelphia USA, 2000.
18/ Gociman B, et al 2004. Expression of angiotenginogen in proximal tubule as a function of glomerular filtration rate. Kidney Int 65: 2153 – 2160.
19/ Boer WH, Braam B. Side effects of reduced renal perfusion pressure and acute volume expansion on proximal tubule and whole kidney angiotensin II content in the rat. Kidney Int. 1997, 51: 44 – 49.
20/ Rohrwasser A et al. Element of a paracrine tubular renin angiotensin system along the entire rephron. Hyperlension 1999, 34: 1265 – 1274
21/ Dimmelers, Rippmann V. Angiotensin II induces Apptosis of Human endo thelial cells. Circulation reseach 1997; 81: 970. 976.
22/ Li D, Yang B, Phillips MI. Proapoptotic effects of ANG II in humam conronary endothelial cells: role of AT 1 receptor and PKC activation. Am J physiol 1999 Mar; 276 (3pt2): 4786 – 92.
23/N.Werner and G. Nickenig. AT1 receptors in atherosclerosis: biological effects including growth, angiogenesis, and apoptosis. European Heart Journal Supplements (2003) 5 (Supplement A), A9-A13
24/ Kathy K. Griendling. NAD(P)H Oxidase Role in Cardiovascular Biology and Disease. Circ. Res. 2000;86;494-501.
25/ Patel RP, Moellering D, Murphy-Ullrich J. Cell signaling by reactive nitrogen and oxygen species in atherosclerosis. Free Radic Biol Med 2000; 28: 1780-94.
26/ Pueyo ME, Gonzalez W, Nicoletti A.Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol 2000; 20: 645-51.
27/Usui M, Egashira K, Tomita H et al. Important role of local angiotensin II activity mediated via type 1 receptor in the pathogenesis of cardiovascular inflammatory changes induced by chronic blockade of nitric oxide synthesis in rats. Circulation 2000; 101: 305-10.
28/ Luft FC, Mervaala E, Muller DN et al. Hypertension-induced end-organ damage. A new transgenic approach to an old problem. Hypertension 1999; 33: 212-8.
29/ Hiroshi Miyazaki, Hidehiro Matsuoka, John P. Cooke. Endogenous Nitric Oxide Synthase Inhibitor : A Novel Marker of Atherosclerosis. Circulation 1999;99;1141-1146.
30/ Isrer JM, Kearney M. Apptosis in human atherosclerosis and restenosis. Circulation 1995; 91: 2703-2711.
31/ Knockx MM. Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol. 1998: 18: 1519-1522.
32/ Libby P. Current concepts of the pathogenesis of the acute coronary syrdromes. Circulation 2001; 104: 365-372.
33/ Marco Valgimigli, Laura Agnoletti. Serum from patents with acute cororary syrdromes displays a proapoptotic effect on Humam endothelial cells; A possible link to Pa-Coronary syndromes. Ciculation, 2003; 107: 264-270.


{kind=link}