

Tóm lược: Về mặt lịch sử, tác dụng đầu tiên được nhắc đến của thuốc ức chếmen chuyển là tăng hoạt tính của Bradykinin, một trong các cơ chất của men chuyển angiotensin (ACE).
Stefano Taddei, L.Bortolotto
Người dịch: Bs. Nguyễn Quang Trung – Bệnh viện Nhân dân Gia Định
Hiệu đính: TS. BS HồHuỳnh Quang Trí – Phó chủtịch Hội Tim mạch TP.HCM
Tuy nhiên trong những năm sau đó, các mô hình phân tử mô tả cơ chế tác dụng của các thuốc ức chế men chuyển trong việc hạ huyết áp lại tập trung chủ yếu vào hệthống renin- angiotensin. Tuy vậy, trong 20 năm gần đây tầm quan trọng của bradykinin trong việc điều hòa dãn mạch, lợi niệu, chống oxy hóa, ly giải sợi, phản ứng viêm và chết theo chương trình ngày càng rõ hơn. ACE có tính gắn kết với bradykinin mạnh hơn với angiotensin I, vì vậy các thuốc ức chếmen chuyển ức chếsựthoái gián bradykinin hiệu quảhơn là ức chế tổng hợp angiotensin II. Các dữliệu mô tả hiệu quảcủa ức chếmen chuyển trên tín hiệu bradykinin ủng hộgiảthuyết là lợi ích bảo vệtim mạch của thuốc ức chếmen chuyển là do tăng bradykinin hơn là do giảm angiotensin II, đặc biệt khi các thuốc này được sữ dụng ở liều cao. Thực vậy, mục tiêu của ức chếmen chuyển là hướng đến việc điều tiết bradykinin trong tế bào nội mô. Các khái niệm mới về mặt cơ chế này có thể dẫn đến sự phát triển những chiến lược tăngtín hiệu bradykinin.
Những điểm mấu chốt:
Các thuốc ức chếmen chuyển có tác động bảo tồn bradykinin phụthuộc liều.
Các lợi ích tim mạch của các thuốc ức chếmen chuyển được ghi nhận khi dùng liều cao. Điều này gợi ý một vai trò quan trọng của bradykinin.
Có sựkhác nhau giữa các thuốc ức chếmen chuyển trong tính chọn lọc trên bradykinin/angiotensin I trong các nghiên cứu in vitro.
1. Giới thiệu
Hơn 30 năm qua, có nhiều nghiên cứu về việc sửdụng các thuốc ức chếhệthống renin-angiotensin trong việc điều trịtăng huyết áp. Cảcác thuốc ức chếmen chuyển và chẹn thụthểangiotensin (ARB) đã chứng minh làm giảm huyết áp, giảm nguy cơ đột quỵ và các triệu chứng của suy tim [1]. Tuy nhiên sựkhác biệt giữa hai nhóm thuốc này trong việc làm giảm nguy cơ tim mạch tồn dư ởbệnh nhân tăng huyết áp ngày càng rõ rệt trong các năm qua. Vào năm 2006, Straus và Hall đã báo cáo kết quảmột phân tích gộp lớn cho thấy các thuốc ức chếmen chuyểnlàm giảm nguy cơ nhồi máu cơ tim 14% (p < 0,00001) so với các ARB làm tăng nguy cơ nhồi máu cơ tim 8% (p = 0,03) [2]. Gần đây, Van Vark và cộng sựđã chứng minh các liệu pháp điều trịcó sửdụng ức chếmen chuyển làm giảm tửvong đáng kể(-10%), trong khi điều trịcó sửdụng ARB không ảnh hưởng dến kết cục tửvong. Một phân tích gộp gồm 195.267 bệnh nhân so sánh ảnh hưởng của các thuốc ức chếmen chuyển và ARB lên các biến cốtim mạch chính so với giảdược [4]. Với cùng mức hạhuyết áp tâm thu/huyết áp tâm trương là 10/5 mmHg, việc điều trịcó sửdụng ức chế men chuyển làm giảm 35% các biến cốmạch vành [khoảng tin cậy (CI) 95%: -52 đến -9], giảm 48% nguy cơ đột ngụy (95% CI -66 đến -19], giảm 53% nguy cơ suy tim (95% CI -74 đến -21], trong khi điều trịvới các ARB hiệu quảgiảm nguy cơ đột quỵ và suy tim đạt được ít hơn [-20% (95%CI -30 đến -7) và -25% (95% CI -40 đến -8), và không giảm các biến cốmạch vành (Hình 1) [4].
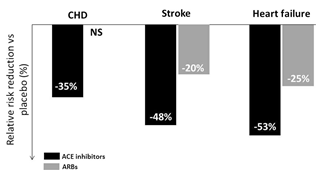
Hình 1: Ảnh hưởng của việc giảm huyết áp bởi ức chếhệthống renin-angiotensin lên việc làm giảm các biến cốtim mạch. Trong một phân tích gộp được công bố gần đây, gồm 195.267 bệnh nhân đánh giá hiệu quảcủa các thuốc ức chế men chuyển và ARB lên các biến cốtim mạch chính [4]. Với cùng mức giảm huyết áp 10/5 mmHg, thuốc ức chế men chuyển làm giảm 35% các biến cốmạch vành, giảm 48% đột quỵ, và giảm 53% suy tim so với giảdược, trong khi đó các ARB không có lợi ích lên các biến cốmạch vành, giảm được 20% đột quỵ và giảm 25% suy tim so với giảdược.
Tuy nhiên trong thửngiệm ONTARGET, nghiên cứu đối đầu lớn nhất giữa thuốc ức chếmen chuyển và ARB, so sánh hiệu quảliều cao của telmisartan 80 mg và ramipril 10 mg trong việc điều trịtăng huyết áp đã chứng minh hiệu quảkhông thua kém của ARB so với ức chếmen chuyển trong bảo vệtim mạch. Một phân tích dưới nhóm theo dõi huyết áp lưu động cho thấy telmisartan giảm huyết áp tâm thu 24h mạnh hơn và có sựkhác biệt 4,1 mmHg vềhuyết áp tâm thu ban đêm. Mặc dù có sựkhác biệt trong mức hạáp, nhưng telmisartan không có được lợi ích tim mạch như ramipril [5].
Cơ chế hoạt động của các thuốc ức chếmen chuyển và ARB chủyếu tác động lên hệthống renin-angiotensin (RAS). Với ức chếmen chuyển, mức angiotensin I (Ang I) tăng, trong khi đó mức angiotensin II (Angio II) giảm, do đó làm giảm giữmuối thông qua Angio II, co mạch, tăng trưởng mạch máu và các ảnh hưởng đông máu (Hình 2)[6]. Các ARB tác động lên cùng một đường qua việcchẹn thụthểAT1 của Angio II. Tuy nhiên cần nhớrằng thuốc ức chếmen chuyển đầu tiên là một peptide được chiếc xuất từđộc tốcủa một loại rắn Brazil có tên là Bothrops jararaca có tác dụng ức chếmạnh mẽsựthoái gián của bradykinin [7]. Enzym này cũng có tác dụng lên chuyển hóa Angio I thành Angio II [8]. Việc phát hiện ACE này vào năm 1970 [9] giúp tìm ra captopril, thuốc ức chếmen chuyển đầu tiên dạng uống.
Tuy nhiên, tầm quan trọng của bradykinin ngày càng trởnên rõ ràng hơn suốt 20 năm qua. Chuyển hóa bradykinin từdạng nonapeptide thành dạng peptide bất hoạt có thểdiễn ra ởhai vùng xúc tác chuyển hóa tương tựcủa ACE, trong khi đó việc chuyển hóa Angio I thành Angio II chỉdiễn ra ởvùng C-terminal cùa ACE [11, 13]. Theo bảng giá trịIC50, bradykinin có kích thước 1,3 µM, Angio I có kích thước 2,7µM, vì vậy bradykinin có khảnăng gắn kết với ACE cao hơn Angio I [14]. Các thuốc ức chếmen chuyển ức chếsựthoái gián bradykinin hiệu quảhơn ức chếsựchuyển hóa tạo Angio II. Chính khảnăng của các thuốc ức chếmen chuyển, không có ở các ARB, làm tăng tín hiệu của bradykinin có những ứng dụng quan trọng vì bradykinin đã được chứng minh là đóng vai trò trung gian trong nhiều hiệu ứng pleiotropic giúp cải thiện trương lực và cấu trúc mạch máu, tăng khảnăng ly giải huyết khối, và bảo vệmạch máu chống lại sựkết tập tiểu cầu và bạch cầu được hoạt hóa [15,17].
Nhiều bằng chứng cho thấy bradykinin có ảnh hưởng lên hệthống tim mạch và lớp nội mô. Trong bài này, chúng tôi nói vềcác bằng chứng ảnh hưởng của bradykinin lên hệthống tim mạch và các ảnh hưởng của ức chếmen chuyển lên bradykinin. Thật vậy, các bằng chứng ủng hộgiảthuyết hiệu quảbảo vệtim mạch của thuốc ức chếmen chuyển là do tăng tác dụng bradykinin hơn là do giảm tác dụng của Angio II.
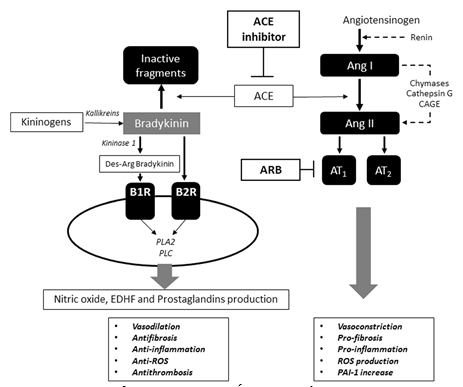
Hình 2: Cơ chế tác dụng của các thuốc ức chếmen chuyển, ARB và bradykinin. Các thuốc ức chếmen chuyển và ARB làm giảm tác dụng Angio II bằng cách ức chếhệthống renin-angiotensin: các thuốc ức chếmen chuyển ngăn tổng hợp Angio II từ Angio I, trong khi đó các ARB ngăn Angio IIgắn vào các thụ thể của nó. Bằng cách làm giảm hoạt tính Angio II, các ức chếhệthống renin-angiotensin làm giảm co thắt mạch, sợi hóa, viêm, sản xuất ROS và mức PAI-1. Hơn nữa, khi ức chếACE là enzym chuyển hóa bradykinin thành dạng bất hoạt, mức bradykinin máu sẽ tăng. Bradykinin thông qua hai thụthểB1R và B2R làm tăng oxide nitric, EDHF và prostaglandin có tác dụng dãn mạch và chống sợi hóa, chống viêm, chống ROS và chống huyết khối. ACE angiotensin converting enzyme, Ang angiotensin, ARB angiotensin receptor blocker, AT1 angiotensin II receptor type I, AT2 angiotensin II receptor type 2, B1R bradykinin receptor 1, B2R bradykinin receptor 2, CAGE chymostatin-sensitive angiotensin II generating enzyme, EDHF endothelium-derived hyperpolarizing factor, PAI-1 plasminogen activator inhibitor-1, PLA2 phospholipase 2, PLC phospholipase C, ROS reactive oxygen species.
2.Bradykinin và các thụthể
ACE ảnh hưởng đến tác động của bradykinin lên hệthống tim mạch, chuyển hóa bradykinin từdạng không peptide thành dạng BK(1-7) heptapeptide và BK(1-5) pentapeptide [18-20]. Pentapeptide BK(1-5) là một chất chuyển hóa ổn đinh, được xem như chất đánh dấu của bradykinin, có thời gian bán hủy (half-life) < 30h [18,20]. Các chất chuyển hóa khác như aminopeptidase, carboxypeptidase và kinase I chuyển bradykinin thành BK(2-9), BK(2-8) đóng vai trò thứyếu trong chuyển hóa bradykinin trong điều kiện sinh lý bình thường [19,22].
Hai thụthểbradykinin B1R và B2R có tác động lên hệthống huyết động nội mô tại chỗ. B2R hiện diện nhiều trong nội mô, trong khi đó B1R có ít và được điều tiết tăng lên trong sựđáp ứng với stress như tình trạng thiếu máu/tổn thương tái tưới máu, viêm mạn hoặc đái tháo đường {23-25]. Các cytokine trợ viêm, các endotoxin, các chất oxy hóa tựdo có tác dụng làm tăng B1R [23,25,26]. Sựchuyển dịch của B1R và B2R là một dấu hiệu của bệnh, như ởbệnh nhân hội chứng mạch vành cấp tỉlệB1R/B2R lớn hơn ởngười khỏe mạnh [27]. Tương tựởloài chuột có đái tháo đường, B1R tăng gấp 3 lần, trong khi đó B2R vẫn không đổi so với chuột không có đái tháo đường [28].
Trong các mô hình thực nghiệm, B2R được hoạt hóa trực tiếp bởi bradykinin, trong khi dó B1R được hoạt hóa bởi chất chuyển hóa Des-Arg(9)-BK [29]. Sựhoạt hóa thụthểgây ra sựdịch chuyển phospholipase A2 tương bào đến màng tếbào, làm tăng Ca2+ nội bào, và phóng thích các chất như nitric oxide (NO), yếu tốtăng hoạt hóa mô (EDHF), prostaglandins và acid epoxyeicosatrienoic (EETs). Các chất này có tác dụng lợi niệu, dãn mạch và giảm stress oxy hóa, chết tếbào theo chương trình, viêm và tiêu sợi huyết. Trong tình trạng khỏe mạnh, B2R được xem là chất điều tiết chính đối với các đáp ứng này. Tuy nhiên các bằng chứng cho thấy trong tình trạng bệnh lý, B1R có thểđiều tiết các phản ứng này [29].
2.1. Tác động lên mạch máu và huyết áp
Trong điều kiện bình thường, bradykinin có tác dụng hạáp [30], giúp ổn định tình trạng huyết động, cân bằng tác động bất lợi của Angio II. Ởchuột được chuyển đổi gen, lượng B2R quá nhiều dẫn đến giảm huyết áp nặng, tác dụng này bịloại bỏbằng cách cho vào chất đối kháng B2R HOE 140 [30]. Cũng trong thửnghiệm này, chuột có chếđộăn nhiều muối, B2R -/- có mức huyết áp tâm thu và áp lực động mạch trung bình cao hơn chuột có B2R +/+ [31,32]. Việc sốlượng B1Rtăng nhiều hoặc mất chức năng không ảnh hưởng đến sựthay đổi huyết áp [33,34] .
Một phân tích gộp gồm 11 thửnghiệm lâm sàng đã chứng tỏvai trò của bradykinin trong việc điều hòa huyết áp, việc giảm B2R có liên quan đến nguy cơ cao tăng huyết áp [35]. Một vài nghiên cứu nhỏcho thấy mối liên quan giữa B1R và tăng huyết áp [37,38]
2.1.1. Dãn mạch
Hiệu quảhạáp của bradykinin là kết quảcủa của dãn mạch toàn thân, dãn mạch thận và hiệu quảlợi niệu [39,40]. Trong các thửnghiệm lâm sàng, khi tiêm bradykinin vào trong động mạch cánh tay sẽlàm tăng dãn mạch và dòng chảy tĩnh mạch có phụthuộc liều dùng[39,40]. Ởmức độphân tử, dãn mạch thông qua hai đường riêng biệt: sựphóng thích NO và prostaglandin và tăng khửcực màng thông qua EETs [19,41]. Trong một thửnghiệm năm 2013, bradykinin gây dãn các động mạch vành bò được gây co thắt chủđộng trước thửnghiệm, hiệu quảdãn mạch của bradykinin bịgiảm bằng cách ức chếtổng hợp nitric oxide (NOS) và ức chếtổng hợp prostaglandin [19]. Hiệu quảdãn mạch này tăng lên khi sửdụng miconazole, một chất ức chếtổng hợp EET [19]. Hiệu quảdãn mạch của bradykinin bịức chếbởi chất đối kháng B2R, không bịảnh hưởng bởi chất đối kháng B1R [19].
Sựkhác biệt này thấy được ởnhững bệnh nhântăng huyết áp và có mức huyết áp bình thường [41]. Ởnhững bệnh nhân có huyết áp bình thường, ức chếNOS gây giảm hiệu quảdãn mạch, nhưng ảnh hưởng khi ức chếEDHF [4]. Ởnhững bệnh nhân tăng huyết áp, kết quảthì ngược lại: đáp ứng dãn mạch của bradykinin phụthuộc vào EDHF [41].
2.1.2. Lợi niệu và chức năng thận
Bradykinin có liên quan đến việc cân bằng muối và B2R [31,32]. Ởchuột có chếđộăn nhiều muối, B2R-/- có mức huyết áp tâm thu và áp lực động mạch trung bình cao hơn ởchuột có B2R -/- và chếđộăn muối bình thường [31,32]. Hơn nữa chuột có B2R -/- duy trì chếđộăn nhiều muối gây tăng co thắt mạch máu thận và giảm lưu lượng đến cách đáng kểkhi được so sánh với chuột có B2R -/- và có chếđộăn muối bình thường [32].
Bradykinin có liên quan đến sựtái hấp thu muối, có ảnh hưởng trực tiếp đến hoạt động các kênh sodium thượng bì ởnephron đoạn xa [42]. Ởchuột có B2R -/-/B1R -/- hoạt tính kênh sodium thượng bì tăng cao đáng kểởchuột hoang có chếđộăn nhiều muối và thấp hơn đáng kểởnhóm chuột hoang có chếđộăn tiết chếmuối [43]. Bradykinin ức chếhoạt động kênh sodium bằng cách giảm khảnăng mởcủa kênh, có phụthuộc liều. Hiệu quảnày giảm đáng kểbởi chất đối kháng B2R với HOE 140 [44]. B2R có vai trò điều hòa hoạt động kênh sodium thượng bì ởống liên kết và ống thu thập [45].
Bằng chứng cho thấy mối liên quan giữa bradykinin và chức năng thận xuất phát từcác nghiên cứu đánh giá mối liên quan giữa kiểu gen B1R và B2R với sựtiến triển của rối loạn chức năng thận [46-48]. Thông qua chuột có tính nhạy cảm muối Dahl người ta chứng minh mối liên quan giữa B2R và chức năng thận, với việc thận bịtổn thương do việc quá tải muối. Việc phóng thích Kallikrein do adenovirus giúp đảo ngược quá trình phì đại cầu thận do muối, làm giảm phản ứng viêm, giảm xơ hóa thận [49]. Những ảnh hưởng này bịloại bỏbởi HOE 140 – chất đối kháng B2R [49]. Sau cùng, sau các tổn thương thận do thiếu máu/ tái tưới máu, sựtổn thương DNA và chết theo chương trình tăng cao đáng kểởchuột có B2R -/-/B1R -/- so với loại chuột hoang dã [50].
Bằng chứng chỉra sựkhiếm khuyết thông qua bradykinin do rối loạn gen hay bệnh lý sẽdẫn đến co thắt mạch và tích lũy muối trong cơ thểđược khảo sát ởcác bệnh lý tăng huyết áp và tim mạch.
2.2. Các ảnh hưởng có liên quan huyết áp
2.2.1. Stress oxy hóa
Tăng tỉlệgốc oxy hóa tựdo/ chất chống oxy hóa gặp ởbệnh nhân tăng huyết áp, stress oxy hóa dẫn đến tổn thương nội mô thận và mạch máu [51]. Bradykinin có thểlàm giảm tình trạng stress oxy hóa ởnội mô bằng cách phóng thích NO [52-54]. Các tếbào nội mô ởngười, NO được tạo ra nhanh và nhiều bởi bradykinin khi phản ứng với chất đối kháng B2R và được tạo ra chậm và ít bởi Des-Arg(9)-BK khi phản ứng với chất đối kháng B1R [52].
CảB2R và B1R đêu có vai trò điều hòa stress oxy hóa. Ởchuột có B1R -/- và chuột có B2R -/- có tình trạng stress oxy hóa tăng cao [55]. Ởchuột có B2R -/- có hàm lượng chất chống oxy hóa vitamin C trong huyết tương thấp hơn so với chuột hoang, cũng như chuột có B1R -/- có gốc oxy hóa tựdo ascorbyl tăng cao trong huyết tương so với chuột hoang [55]. Khi các chuột này được đưa vào trong mô hình thực nghiệm tổn thương thận do thiếu máu/ tái tưới máu, tổn thương DNA và chết tếbào theo chương trình do chất oxy hóa cao nhất trong nhóm chuột B2R -/-/B1R -/-, kếđó là B2R -/- và cuối cùng là nhóm chuột hoang [50].
2.2.2. Ly giải fibrin
Sựcân bằng ly giải fibrin quyết định sựhình thành và thoái gián cục máu đông, được duy trì bởi sựcân bằng giữa yếu tốhoạt hóa plasminogen mô (t-PA) và ức chếyếu tốhoạt hóa plasminogen mô loại 1 (PAI-1) [56]. Việc phá vỡdẫn đến tăng PAI-1 hoặc giảm t-PA liên quan đến việc hình thành cục huyết khối và tạo ra yếu tốnguy cơ cho bệnh tim mạch [57,58]. Các bằng chứng cho thấy cảhai hệthống RAS và bradykinin có liên quan đến sựcân bằng này và sựtương tác giữa hai hệthống này ảnh hưởng đến nồng độhuyết tương của t-PA và PAI-1 [56,59].
Việc tiêm bradykinin vào trong động mạch cánh tay của những người khỏe mạnh không có tăng huyết áp dẫn đến tăng nồng đột-PA, có phụthuộc liều dùng [40,60,61]. Ởnhững bệnh nhân tăng huyết áp, mặc dù lượng NO có giảm [62], bradykinin vẫn thúc đẩy phóng thích t-PA bằng cách hoạt hóa đường dựtrù có liên quan đến phức hợp nhạy cảm sulphaphenazol [63]. Rõ ràng bradykinin vẫn duy trì một sốkhảnăng như một chất chống đông bằng cách hoạt hóa con đường trù bị.
Hiệu quảlên PAI-1 thì ít rõ ràng. Một sốnghiên cứu ởngười khỏe mạnh và bệnh nhân tăng huyết áp [39,60,64]. Tiêm bradykinin vào trong cánh tay không ảnh hưởng đến mức PAI-1 huyết tương [40]. Ởtếbào nội mô động mạch chủchuột, bradykinin làm tăng mức biểu hiện protein và ARN thông tin (mARN) của PAI-1, hiệu quảnày bịức chếbởi HOE – chất đối kháng B2R [65].
2.2.3. Phản ứng viêm và chết tếbào theo chương trình
Trong nhồi máu cơ tim, tổn thương do thiếu máu cục bộ – tái tưới máu dẫn đến phản ứng viêm và chết tếbào nội mô theo chương trình, thúc đẩy dẫn đến xơ vữa mạch và các biến cốtim mạch trong tương lai [66]. Bệnh thận và sựtiến triển của bệnh được đặc trưng bởi phản ứng viêm và chết tếbào theo chương trình. Bradykinin được chứng minh làm giảm phản ứng viêm và chết tếbào theo chương trình trong các bệnh lý này.
Trong mô hình thực nghiệm tổn thương tim do thiếu máu cục bộ – tái tưới máu ởthỏ, việc bổsung bradykinin giúp làm giảm mức các cytokine phản ứng viêm và sốtếbào cơ tim chết theo chương trình [67]. Hiệu quảcủa bradykinin lên chết tếbào theo chương trình bịức chếbởi chất đối kháng bradykinin và ức chếtổng hợp NO [67]. Tương tựtrong mô hình thực nghiệm rối loạn chức năng thận, việc tiêm lâu dài bradykinin vào chuột nhạy cảm muối Dahl có chếđộăn nhiều muối giúp bảo vệthận chống lại tổn thương thận do muối, tăng mức NO, và giảm sựtích tụcủa đại thực bào/đơn bào trong gian mô, chết theo chương trình tếbào ống thận, tăng sinh oxi hóa, mức yếu tốtăng trưởng β[68]. Trong mô hình và liều dùng này, mức huyết áp không bịảnh hưởng, do đó ảnh hưởng lên phản ứng stress oxy hóa, phản ứng viêm, và chết tếbào theo chương trình có thểxảy ra độc lập với huyết áp.
3. Bradykinin trong quá trình lão hóa và bệnh lý tim mạch
Nhiều báo cáo cho thấy giảm đáp ứng bradykinin trong các mô hình thực nghiệm lâm sàng. Động mạch chủcủa các con chuột già giảm đáp ứng với kích thích bradykinin từbên ngoài [69]. Đáp ứng giảm huyết áp khi truyền liều bradykinin sinh lý ởchuột 5 tháng tuổi cao gấp bốn lần so với chuột 18 tháng tuổi. Điều này được giải thích là do sựgiảm B2R bradykinin có liên quan đến tuổi được khảo sát ởcác mô hình chuột khác nhau [70]. Trong nuôi cấy, những tếbào tim lão hóa mất dần khảnăng bộc lộB2R, sựmất khảnăng này xảy ra đồng thời với mất khảnăng tân tạo mạch máu mới của những tếbào lão hóa [71]. Một nghiên cứu phân tửởngười cho thấy B2R tăng ởnhững người bình thường cho đến 50 tuổi. Tuy nhiên những bệnh nhân có bệnh mạch vành, B2R giảm nhanh ờtuổi từ50 đến 65 [72]. Sựảnh hưởng của bradykinin ởngười giảm dần theo tuổi xảy ra đồng thời với giảm dần ảnh hưởng dãn mạch do nội mô [73,74].
Các kích thích bệnh lý như tăng huyết áp thúc đẩy quá trình lão hóa liên quan đến bradykinin. Mức kininogen và các yếu tốtạo kinin giảm ởnhững bệnh nhân tăng huyết áp, do sựsản xuất các tiền chất trọng lượng phân tửcao của kinin ởgan bịgiảm [75]. Trong phân tích dưới nhóm PERTINENT của nghiên cứu EUROPA, mức bradykinin giảm 30% ởnhóm bệnh nhân có bệnh mạch vành so với người khỏe mạnh (12,4 sao với 18,3 pg/mL; p < 0,001) [76]. Khảnăng bộc lộvà hoạt tính của B2R giảm có liên quan theo tuổi, mức bradykinin giảm ởnhững bệnh nhân tăng huyết áp và bệnh mạch vành do mức kinin lưu hành trong máu giảm.
4. Các thuốc ức chếmen chuyển và bradykinin
Ởnhững bệnh nhân tăng huyết áp có sựmất cân bằng giữa RAS (chủyếu Angio II) và bradykinin. Mức bradykinin giảm trong khi đó Angio II thì tăng cao [75]. Các thuốc ức chế men chuyển,qua cạnh tranh với bradykinin ở vịtrí gắn ACE, làm giảm sựthoái gián bradykinin, dẫn đến tăng nồng độbradykinin trong mô và huyết tương [77]. Trong một nghiên cứu trên các mẫu tế bào nội mô và đoạn động mạch chủ, việc điều trịvới Perindopril giúp làm giảm sựthoái gián bradykinin thành BK (1-5) ở49% mô khỏe mạnh và 76% mô từchuột tăng huyết áp tự nhiên 4 tháng tuổi [78]. Những bệnh nhân suy tim được điều trịvới perindopril và enalapril ghi nhận tăng từbốn đến mười lần mức bradykinin huyết tương và hai lần mức BK(1-7) huyết tương [79]. Mặc dù bradykinin không tác dụng trực tiếp lên B1R, nhưng chất chuyển hóa của bradykinin Des-Arg(9)-BK có tác động. Với ức chếmen chuyển, càng nhiều bradykinin thì càng nhiều Des-Arg(9)-BK được chuyển hóa bởi kininase I [22].
Các thửnghiệm lâm sàng đã chứng minh bradykinin có liên quan đến hiệu quảchống tăng huyết áp của thuốc ức chếmen chuyển [80,81]. Ởnhững bệnh nhân có huyết áp bình thường, hiệu quảhạhuyết áp của việc điều trịbằng captopril và perindopril giảm khi dùng kèm chất đối kháng B2R- HOE 140 [80,81]. Ởnhững bệnh nhân tăng huyết áp, việc điều trịbằng lisinopril giúp tăng dãn mạch và tăng dòng máu chảy ởcánh tay khi truyền bradykinin [82]. Tương tựcác nghiên cứu ởnhững bệnh nhân suy tim, ức chếmen chuyển làm tăng sựdãn mạch và dòng máu chảy ởcánh tay khi truyền bradykinin [83,84]. Hiệu quảnày bịức chếkhi dùng đồng thời HOE 140, nhưng không phải đồng vận hay chất đối kháng B1R [84]. Cuối cùng một nghiên cứu vềkiểu hình B2R cho thấy tăng dòng máu chảy cánh tay và giảm trởkháng mạch máu cánh tay khi truyền đồng thời bradykinin/enalaprilat nhiều hơn đáng kểởnhững bệnh nhân có kiểu gen BEI -9/-9 so với kiểu gen BEI +9/+9 [36].
Ức chếmen chuyển giúp tăng cường hiệu quảhạáp của bradykinin. Mối liên quan của bradykinin và ức chếmen chuyển trong đáp ứng nội mô được chứng minh trong phân tích dưới nhóm PERTINENT của nghiên cứu EUROPA, gồm 1200 bệnh nhân bệnh mạch vành ổn định. Sau 1 năm điều trịvới perindopril 10 mg, mức bradykinin tăng đáng kểso với giảdược (p< 0,001) (Hình 3) [76]. Một điều thật thú vị, perindopril 10 mg giúp phục hồi bradykinin vềmức bình thường tương tựnhư những người tình nguyện. kèm theo giảm 31% chết tếbào theo chương trình ởmô (p<0,05), tăng 19% protein NOS nội mô (p<0,05), và tăng 27%hoạt tính NOS nội mô (p<0,05). Ởnhững bệnh nhân này khi dùng đồng thời chất đối kháng HOE 140 của B2R làm giảm ảnh hưởng lên NOS nội mô và chết tếbào theo chương trình [76]. Điều thú vịlà lợi ích này thấy được trong thửnghiệm EUROPA thì đồng thời cũng thấy được trong những thửnghiệm lâm sàng khác với perindopril [85].
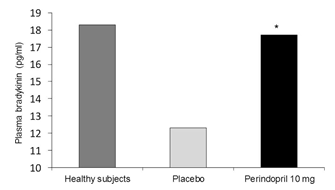
Hình 3: Hiệu quảlâu dài của perindopril 10 mg lên mức bradykinin. Trong phân nhóm PERTINENT của thửnghiệm EUROPA, những bệnh nhân được điều trịvới perindopril 10 mg có mức bradykinin phục hồi tương tựnhư những người tình nguyện. *P < 0,05 cho sựcải thiện khi so với ban đầu khi dùng perindopril 10 mg so với giảdược. Đã được chấp thuận [76].
Ảnh hưởng của ức chếmen chuyển lên tiêu sợi huyết được chứng minh trong một nghiên cứu gồm 16 người lớn có huyết áp bình thường được truyền bradykinin vào động mạch cánh tay. Sựphóng thích t-PA hoạt hóa từnội mô sau đáp ứng kích thích tại chỗcủa bradykinin tăng lên hơn gấp đôi khi được điều trịquinapril so với được điều trịbằng giảdược hoặc losartan [86]. Tương tựtrong một nghiên cứu ởnhững bệnh nhân suy tim có triệu chứng, liệu pháp ức chếmen chuyển gây tăng phóng thích t-PA tại chỗsau khi truyền bradykinin vào động mạch cánh tay {83,84]. Tác dụng này bịloại bỏbởi HOE 140, mà không phải chất đối kháng B1R [84]. Tác dụng nhóm cũng đã chứng minh khi điều trịnhững bệnh nhân đái tháo đường có tăng huyết áp với perindopril giúp làm giảm mức PAI-1 huyết tương [87]. Giảm PAI-1 không thấy ởlosartan mặc dù có mức hạhuyết áp tương tự, hiệu quảnày do ảnh hưởng thông qua tác động của bradykinin hơn là tác động của Angio I/Angio II [87].
Mô hình thực nghiệm ởđộng vật có tăng huyết áp và bệnh lý tim mạch đã giúp củng cốcác phát hiện lâm sàng này. Trong một nghiên cứu ởnhững con chuột tăng huyết áp tự nhiên, khi sửdụng HOE 140 làm mất đi khảnăng gây tăng tạo GMP vòng ởđộng mạch chủ, một chất có tác dụng gây dãn mạch, do tác động của ramipril và perindopril [88]. Ởchuột B1R -/-/B2R -/-, việc điều trịbằng captopril giúp làm giảm hoạt động kênh sodium thượng bì mạnh hơn đáng kểkhi so với ởchuột hoang [43]. Ởchuột Wistar có đái tháo đường khi điều trịbằng perindopril hoặc ramipril giúp làm giảm tăng huyết áp và trọng lượng mạch máu mạc treo ruột và tỉlệlớp áo giữa: diện tích lồng ống mạch [89]. Dùng đồng thời chất đối kháng HOE 140 của B2R tiêm dưới da làm mất đi tác dụng của các thuốc ức chếmen chuyển lên trọng lượng mạch và tỉlệlớp áo giữa: diện tích lòng mạch [89]. Tương tựởchuột có đái tháo đường khi sửdụng ramipril giúp ngăn chặn albumin niệu và xơ hóa cầu thận [90]. HOE 140 làm mất đi các tác dụng này.
Ức chếmen chuyển thông qua thụthểB1R mang đến những lợi ích khác. Trong thửnghiệm EUROPA gồm những bệnh nhân với bệnh mạch vành ổn định, sựđa dạng kiểu hình của B1R có liên quan đến việc làm giảm kết cục chính (tửvong do nguyên nhân tim mạch, nhồi máu cơ tim không tửvong, và ngưng tim được cứu sống) khi được điều trịvới perindopril 10 mg [91]. Ức chếmen chuyển giúp hoạt hóa B1R cũng được chứng minh ởnội mô [22,92]. Một nghiên cứu trên động mạch vành của bò bị co thắt từ trước cho thấy điều trịcaptopril giúp tăng cường mức dãn mạch thông qua bradykinin, hiệu quảnày bịức chếbởi chất NOS và chất đối kháng B1R, không phải chất đối kháng B2R [19]. Hơn nữa việc điều trịvới enaprilat giúp tăng hoạt hóa trực tiếp B1R, dẫn đến tăng phóng thích NO ởtếbào nội mô [93]. Một sốức chếmen chuyển hoạt động như đồng vận trực tiếp của B1R [22,93].
Nhiều điều chưa được hiểu hết vềvai trò của B2R và B1R trong những giai đoạn bệnh khác nhau. Trong mô hình thiếu máu cục bộ cơ tim ởchuột, ramipril và đồng vận B2R làm giảm kích thước mảng nhồi máu đáng kể, nhưng đồng vận B1R Des-Arg(9)-BK thì không có hiệu quảnày. Tuy nhiên ởmô hình thực nghiệm chuột có đái tháo đường, không phải ramiprilat cũng không phải đồng vận B2R làm giảm kích thước mảng nhồi máu, mà là Des-Arg(9)-BK làm giảm 44% kích thước mảng nhồi máu [28]. Những dữliệu rồi cho thấy sựphức tạp của B2R/B1R và nhấn mạnh vai trò của bradykinin trong việc bảo vệ tim mạch thông qua tác dụng của thuốc ức chếmen chuyển [26].
Cuối cùng, B2R tăng mức biểu thịởngười trẻ, vì vậy những bệnh nhân trẻsẽhưởng lợi nhiều hơn khi được sửdụng ức chếmen chuyển trong điều trị, tăng mức bradykinin sinh khảdụng sẽgiúp tăng kích thích các thụthểtựdo. Ý kiến này giống với dữliệu được công bốtrong thửnghiệm phân tích gộp năm 2009 của Brugts và cộng sự[85], bao gồm 29.000 bệnh nhân từcác nghiên cứu EUROPA, ADVANCE và PROGRESS. Mặc dù giảm được 18% kết cục các biến cốtim mạch và tửvong trên dân sốchung, nhưng những bệnh nhân dưới 60 tuổi đạt được nhiều lợi ích hơn khi được điều trịvới perindopril, phần lớn được sửdụng liều 10 mg (hình 4). Hơn nữa, trong mô hình tiền lâm sàng chuột có tăng huyết áp, việc sửdụng perindopril thời gian ngắn khi bắt đầu có tăng huyết áp giúp ngăn ngừa sựtăng huyết áp do lão hóa [94]. Những dữliệu này ủng hộý kiến cho rằng các thuốc ức chếmen chuyển có hiệu quảbảo vệmạch máu nhiều hơn ởngười trẻvà khảnăng làm chậm sựtiến triển của bệnh lý tim mạch khi được sửdụng sớm sau chẩn đoán tăng huyết áp.
4.1.Sựkhác biệt giữa các thuốc ức chếmen chuyển
Các thuốc ức chếmen chuyển được phân thành ba nhóm dựa trên nhóm gắn kết chức năng: captopril và zofenopril có chứa gốc sulfhydryl; benazepril, enalapril, lisinopril, perindopril, quinapril, Ramipril, và trandolapril chứa gốc dicarboxylate; và fosinopril chứa gốc phosphonate [95]. Sựkhác biệt vềcấu trúc này ảnh hưởng đến dược động và dược lực của thuốc. Vịtrí gắn kết của ức chếmen chuyển có ảnh hưởng đến hiệu quảthuốc thông qua bradykinin và Angio-I. Trong thửnghiệm vềmức gắn kết in vitro sửdụng tếbào nội mô người, tỉlệchọn lọc gắn kết bradykinin/Ang I là 1.44 đối với perindopril, 1,16 đối với ramiprilat, 1,09 đối với quinaprilat, 1,08 đối với trandoprilat, và 1.00 đối với enaprilat. Dữliệu cho thấy so với các thuốc ức chếmen chuyển khác, perindopril có tính chọn lọc trên bradykinin nhiều hơntrên Ang I [14]. Các thuốc ức chếmen chuyển cũng có mức gắn kết với mô tim khác nhau, cao nhất là perindopril, theo sau là quinalaprilat, ramiprilat, kếđó là enalaprilat và fosinoprilat, và sau cùng là captopril [96].
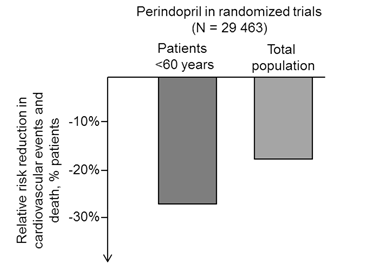
Hình 4: Ảnh hưởng có liên quan đến tuổi của thuốc ức chếmen chuyển perindopril giúp làm giảm tửvong và các biến cốtim mạch trong các thửnghiệm lâm sàng. Trong phân tích gộp từcác thửnghiệm EUROPA, ADVANCE, và PROGRESS, perindopril giúp làm giảm nguy cơ tương đối các biến cốtim mạch và tửvong nổi bật ởnhóm bệnh nhân trẻ(< 60 tuổi) so với nhóm dân sốchung. Sựkhác biệt này được giải thích do mức bradykinin có chức năng ởngười trẻnhiều hơn [85].
Sựkhác biệt vềvai trò sinh học quan trọng của bradykinin chưa được hiểu đầy đủ. Nhờvào sựkết hợp tính ái mỡ, gắn kết cao với mô, và có tỉlệchọn lọc bradykinin/Ang I cao perindopril cho hiệu quảức chếchuyển hóa bradykinin kéo dài hơn, hoàn toàn hơn và do đó tăng hiệu ứng sinh học hơn so với các thuốc ức chế men chuyển khác. Ít nhấtmột nghiên cứu đối đầu đã ủng hộgiảthuyết này và chứng minh các thuốc ức chếmen chuyển có ảnh hưởng khác biệt trên chết tếbào nội mô theo chương trình phản ánh sựkhác biệt trong tỉlệchọn lọc bradykinin/Ang I [14,97]. Khi sửdụng các thuốc ức chế men chuyển cho chuột trong khoảng thời gian 7 ngày với liều hiệu quảhạáp giống nhaugiúp giảm tỉlệchết tếbào nội mô theo chương trình thông qua lipopolysaccharide. So với chất nghiên cứu, perindopril làm giảm hiệu quảkhác biệt có ý nghĩa thống kê (p < 0,001), hiệu quảnày không thấy với các thuốc ức chếmen chuyển khác. Thứtựđộmạnh trong thực nghiệm này là perindopril >ramipril >> quinapril = trandolapril = enalapril. Sựkhác biệt giữa các thuốc ức chếmen chuyểncó ý nghĩa khi so perindopril với quinapril (p < 0,01), perindopril với trandolapril (p < 0,001), và perindopril với enalapril (p < 0,001) [97]. Dữliệu này cho thấy perindopril có hiệu quảcao trên nội mô, tương tựvới thửnghiệm PERTINENT, báo cáo giảm 31% chết tếbào theo chương trình ởtếbào nội mô người, và gợi ý perindopril có khảnăng bảo tồn bradykinin cao ởnội mô [76].
4.2.Vai trò của bradykinin trong đáp ứng liều đối với các ức chếhệthống Renin-Angiotensin
Một bài báo được công bốgần đây trình bày các bằng chứng cho thấynhững khác biệt phụthuộc liều giữa các thuốc ức chếmen chuyển và ARB [98]. Những yếu tố này đặc biệt thú vị để thảo luận trong bối cảnh hiểu biết của chúng ta về bradykinin.
Vềkhía cạnh giảm huyết áp, thuốc ức chếmen chuyển perindopril được chứng minh trong nhiều nghiên cứu có khảnăng làm giảm thêm huyết áp khi tăng từ5 mg lên 10 mg. Trong nghiên cứu Confidence [99], perindopril được tăng liều từ5 mg lên 10 mg sau 2 tuần (N= 1943 và mức huyết áp tâm thu trung bình là 154,5 mmHg), SBP giảm -8 mmHg sau 2 tuần và giảm -18 mmHg sau 12 tuần điều trị(p so với ban đầu < 0,01). Mức giảm huyết áp đáng kểthấy ở193 bệnh nhân có tăng huyết áp nặng lúc ban đầu (mức huyết áp tâm thu trung bình ban đầulà 182,4 mmHg). SBP giảm -23 mmHg với perindopril 5 mg (đo ởtuần thứ2) và giảm -38 mmHg với perindopril 10 mg (đo ởtuần 12). Trong một nghiên cứu gần đây [100], gồm 1004 bệnh nhân tăng huyết áp độ1 và độ2 (mức trung bình SBP/DBP là 155,8/92,8 mmHg), được điều trịvới perindopril 5 mg giảm huyết áp trung bình 9,6/5 mmHg trong 2 tuần, trong khi đó những bệnh nhân được sửdụng perindopril 10 mmHg có mức giảm huyết áp trung bình 18,1/9,5 mmHg. Hiệu quảgiảm huyết áp phụthuộc liều không thấy với ARB. Thật vậy, các ức chếRAS có xu hướng giảm giảm huyết áp nhanh vì nó ngăn chặn việc gắn kết Ang II có tác dụng gây co mạch với thụthểAT1. Tuy nhiên, hiệu quảnày nhanh chóng bịbão hòa, ARB có đường cong đáp ứng liều dạng lõm không sâu, và không có lợi ích lên hiệu quảgiảm huyết áp khi tăng liều từ50% lên 100% [101]. Hơn nữa, cơ chếtác động trực tiếp của ARB có nguy cơ cao hơn gây ra các cơn tụt huyết áp thoáng qua [102]. Xem xét 16 thửnghiệm gồm hơn 113.000 bệnh nhân, việc sửdụng ARB làm tăng nguy cơ tụt huyết áp 56% (p< 0,001). Các tác giảđồng ý với tác dụng phụbất lợi này như là sựgiải thích cho việc tăng tửvong chung do mọi nguyên nhân (+3;p= 0,05) được báo cáo trong nghiên cứu.
Thật vậy, từcơ chếhoạt động mà hiệu quảphụthuộc liều của ARB khá hạn chế. Hoạt động của các ARB dựa trên mức giảm của đường chuyển hóa Ang. Một khi sựbão hòa dược học và sựức chếtoàn phần xảy ra, mức giảm Ang không thểxảy ra hơn nữa và sựtăng liều không có ý nghĩa. Mặc dù tất cảcác thuốc ức chế RAS đều có đặc tính này, các thuốc ức chếmen chuyển làm tăng sinh khảdụng của bradykinin. Nghiên cứu tiền lâm sàng của Cambell và cộng sự[77] đã chứng minh với liều thấp của perindopril, tỉlệAng II/Ang I giảm một cách đáng kể. Tuy nhiên việc tăng bradykinin huyết tương gần như tuyến tính với việc tăng liều perindopril. Do đó, với các ức chếACE, hoạt động bradykinin trởnên ưu thếkhi lợi ích của sựgiảm Ang II bịbão hòa. Hình 5 tóm tắt lại sựkhác biệt mô hình hoạt động của các thuốc ức chếmen chuyển và ARB.
Trong thực hành lâm sàng, sựkhác biệt vềhiệu quảcủa các thuốc ức chếmen chuyển lên Ang II và bradykinin đã được ghi nhận một thời gian dài trước đây. Năm 1986 trong một nghiên cứu trên 18 người đàn ông có huyết áp bình thường, việc ức chếkhảnăng hoạt động của ACE trong huyết tương đểtạo ra Ang II bão hòa, dạng bình nguyên, khi đạt đến liều 10 mg với tỉlệức chếlà 80, 84, 95, và 96% với liều perindopril tương đương là 2,5–5– 10 và 20 mg [103]. Ngược lại, một thửnghiệm ngẫu nhiên, mù đôi chưa được công bốtrước đó được thực hiện ởHeidelberg Australia trên 12 người lớn trưởng thành khỏe mạnh có huyết áp bình thường, khoảng dưới đường cong kéo dài hơn 48 giờchứng tỏmức bradykinin huyết tương khi tăng liều perindopril (Hình 6) [104]. Không có sựbão hòa rõ ràng thấy được ngay cảkhi liều perindopril cao hơn được dùng trong các thực nghiệm lâm sàng. Những bệnh nhân này có tuổi đời trung bình là 27 ±2,8 và cân nặng 72,8 ± 3,5 kg, có mức huyết áp ban đầu SBP/DBP là 124,8 ± 2,2 mmHg, và được sửdụng perindopril liều uống 5, 10, và 20 mg hoặc giảdược ởcác khoảng cách thời gian 7 ngày.
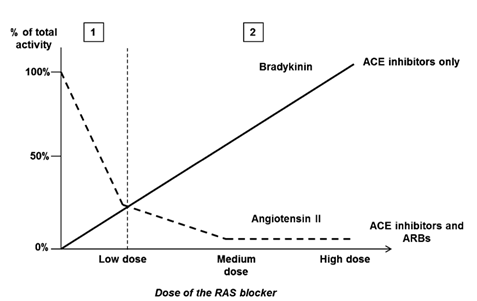
Hình 5: Mô hình dựđoán ảnh hưởng của bradykinin và angiotensin II lên hiệu quảphụthuộc liều của các thuốc ức chếRAS. Ởliều tương đối thấp (1), các thuốc ức chếmen chuyển và ARB làm giảm hoạt tính của angiotensin II. Hiệu quảnày nhanh chóng bịbão hòa. Ởliều cao (2), các thuốc ức chếmen chuyển giúp bảo tồn bradykinin tránh bịthoái gián. ACE angiotensin converting enzyme, ARB angiotensin receptor blocker, RAS renin-angiotensin system
Hiệu quảtăng mức hạáp của bradykinin phụthuộc liều của các thuốc ức chếmen chuyển đi kèm với hiệu quảbảo vệtim mạch thông qua tác dụng của bradykinin [105-107]. Trong thửnghiệm DAPHNET gồm 57 bệnh nhân tăng huyết áp kèm đái tháo đường, liều perindopril được tăng từ5 mg lên 10 mg mỗi ngày khoảng 6 tháng đã cho được những kết quảquan trọng như tăng khảnăng chun dãn động mạch cảnh và đường kính trong động mạch cảnh và chỉsốđo lường độđàn hồi Young có giảm hơn ởnhóm perindopril 10 mg so với nhóm 5 mg. Không có sựkhác biệt vềhuyết áp theo dõi liên tục 24 h được ghi nhận [105]. Phân tích hồi qui xác định những thông sốđộcứng động mạch cảnh có liên quan đến liều perindopril mà không ảnh hưởng đến huyết áp trung bình 24 h [105]. Tương tựtrong thửnghiệm SECURE, một nhóm 732 bệnh nhân với các yếu tốnguy cơ và bệnh lý mạch máu, ramipril 10 mg làm giảm sựtiến triển xơ vữa mạch đáng kểtrong khi đó ramipril 2,5 mg có hiệu quảhạáp tương tựnhưng không làm giảm được tiến triển xơ vữa mạch [107]. Trong một phân tích dưới nhóm của nghiên cứu EUROPA ởnhững bệnh nhân bệnh mạch vành ổn định nguy cơ thấp có huyết áp bình thường lúc vào nghiên cứu (SBP < 120 mmHg), được điều trịvới perindopril 10 mg không làm giảm huyết áp nhưng làm giảm kết cục chính như tửvong do nguyên nhân tim mạch, nhồi máu cơ tim không tửvong, hoặc ngưng tim được cứu sống (giảm nguy cơ tương đối 32%) [106]. Trong những nghiên cứu chứng minh hiệu quảđộc lập với việc hạ huyết áp này, chúng ta có thể đưa ra giảthuyết làlợi ích của việc dùng liều cao thuốc ức chếmen chuyển phản ánh sựtăng bradykinin phụthuộc liều được mô tảởhình 6.
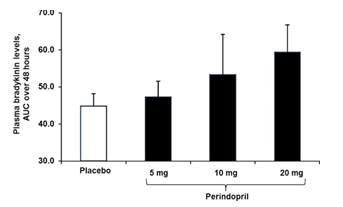
Hình 6: Hiệu quảphụthuộc liều của perindopril. Mức bradykinin huyết tương được đo ở12 người khỏe mạnh có huyết áp bình thường sau khi được điều trịvới perindopril 5, 10, và 20 mg * hoặc giảdược bằng đường uống ởkhoảng cách thời gian 7 ngày [104]. Các mẫu máu được lấy sau 0, 1, 2,4, 8, 24, và 48 h sau mỗi lần uống. Giá trịAUC được tính bằng trung bình ±trung bình sai sốđộlệch chuẩn. * Tương đương với 4, 8, và 16 mg ten-butylamine muối. AUC area under the curve.
Với tính hiệu quảnhóm, khảnăng dung nạp thuốc được quan tâm nhiều hơn khi tăng liều các thuốc ức chếmen chuyển. Tuy nhiên nên nhớrằng các thuốc ức chếmen chuyển là những thuốc chống co mạch có tác dụng phục hồi sựcân bằng giữa Ang II và bradykinin và kiểu hoạt động của các thuốc ức chếmen chuyển là gián tiếp. Trước hết Ang II bịgiảm dần đến mức tối thiểu bởi vì sựsản xuất của Ang II bịgiảm trong khi đó sựthoái gián của Ang II bởi angiotensinase vẫn tiếp tục. Vì thời gian bán hủy của Ang II là 30 phút trong các mô, sựức chếtối đa bởi các thuốc ức chếmen chuyển sẽdần đạt được qua nhiều giờ. Hơn nữa, sựthoái gián bradykinin chậm được phục hồi vềmức sinh lý bình thườngvà chậm đạt đến nồng độcó hiệu quảdãn mạch. Với cơ chếhoạt động này, trong nghiên cứu EUROPA, perindopril 10 mg được dung nạp tốt bất chấp mức huyết áp ban đầu. Không có bất kỳ mối lo ngại nào được báo cáo khi khởi trịcho những bệnh nhân có mức huyết áp tâm thu ban đầu thấp (< 120 mmg) với liều perindopril 10 mg [106].Hơn nữa trong các thửnghiệm có sửdụng các thuốc ức chếmen chuyển, nguy cơ tụt huyết áp được báo cáo là thấp (1,9% với ramipril trong nghiên cứu HOPE [108] và 1% với perindopril trong nghiên cứu EUROPA [109]). Ngược lại, các ARB chỉức chếđược thụthểAT1, có hiệu quảtrực tiếp như đã được thảo luận trước đó, có liên quan làm tăng nguy cơ tụt huyết áp và các biến cốcó hại [102]
Ho khan là một tác dụng không mong muốn khác liên quan với ức chếACE. Trong các thửnghiệm ngẫu nhiên có đối chứng, tỉlệho khan chiếm khoảng 11% theo báo cáo của Physicians’ Desk Reference [110]. Ho khan không phụthuộc vào liều lượng thuốc sửdụng [111]; tuy nhiên cơ chếho khan thì chưa rõ ràng. Trong một nghiên cứu vềmối liên quan giữa sựđa dạng trong kiểu hình của thụthểB2R và nguy cơ ho khan đã kết luận rằng có mối liên quan trực tiếp giữa mức bradykinin và ho khan [112], trong một nghiên cứu khác, thromboxane, một trong những sản phẩm cuối cùng của sựchuyển hóa acid arachidonic thông qua cyclo-oxygenase, được mô tảnhư là chất trung gian của ho khan có liên quan đến ức chếACE [113]. Ngay cảnếu ho khan có liên quan đến bradykinin, thì điều quan trọng cần nhớlà các thuốc ức chếmen chuyển có xu hướng phục hồi bradykinin, thường ởmức thấp ởnhững bệnh nhân tăng huyết áp, vềmức bình thường hơn là làm tăng bradykinin trên mức sinh lý, điều này đã được chứng minh trong nghiên cứu PERTINENT với liều perindopril 10 mg.
5. Kết luận
Nhiều dữliệu nghiên cứu đã giúp làm sáng tỏmối liên quan giữa bradykinin và những lợi ích tim mạch của thuốc ức chếmen chuyển. Cho đến ngày hôm nay, bên cạnh hầu hết các dữliệu dựa trên hiệu quảhạáp và mức huyết tương Ang I/Ang II, còn có các dữliệu quan trọng vềvai trò của hệthống bradykinin. Sựtăng mức bradykinin dường như không phải là hiệu quảcộng thêm của các thuốc ức chếmen chuyển, mà là một thành phần cơ bản trong cơ chếtác dụng của các thuốc này. Tác động đặc hiệu của thuốc ức chếmen chuyển lên bradykinin giải thích sựkhác nhau vềhiệu quảhạ huyết áp và bảo vệtim mạch giữa các thuốc ức chếmen chuyển và các thuốc chẹn thụ thể angiotensin. Ngoài ra, hệthống bradykinin có hoạt tính mạnh mẽởngười trẻ, điều này nhấn mạnh hơn tính hợp lý khi khởi trịnhững bệnh nhân tăng huyết áp với thuốc ức chếmen chuyển.
Tài liệu tham khảo
1.FerrariR.RAASinhibitionandmortalityinhypertension: from pharmacologyto clinicalevidence. KardiolPol.2013;71(1):1–7.
2. Strauss MH, Hall AS. Angiotensin receptor blockers may increaseriskofmyocardialinfarction:unraveling theARB-MI paradox.Circulation.2006;114(8):838–54.
3.vanVarkLC,BertrandM,AkkerhuisKM,etal.Angiotensin-convertingenzymeinhibitorsreducemortalityinhypertension: ameta-analysisof randomizedclinicaltrialsofrenin-an- giotensin-aldosterone systeminhibitorsinvolving158,998 patients.EurHeartJ.2012;33(16):2088–97.
4.Thomopoulos C,ParatiG,ZanchettiA.Effectsofbloodpres- sureloweringonoutcomeincidenceinhypertension:4.Effectsofvariousclassesofantihypertensive drugs–overviewandmeta- analyses.JHypertens.2015;33(2):195–211.
5.ManciaG,ParatiG,BiloG,etal.Ambulatory bloodpressure valuesintheOngoing TelmisartanAloneandinCombination withRamipril GlobalEndpointTrial (ONTARGET).Hyper- tension.2012;60(6):1400–6.
6.Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol.2002;89(2A):3A–9A.
7.FerreiraSH.Abradykinin-potentiatingfactor(Bpf)presentin thevenomofBothropsjararaca.BrJPharmacolChemother.1965;24:163–9.
8.NgKK,VaneJR.Conversion ofangiotensinItoangiotensinII. Nature.1967;216(5117):762–6.
9.YangHY, Erdos EG,LevinY.Adipeptidylcarboxypeptidase thatconvertsangiotensinIandinactivatesbradykinin. Biochim BiophysActa.1970;214(2):374–6.
10.OndettiMA,RubinB,CushmanDW.Designofspecific inhi- bitorsofangiotensin-converting enzyme:newclassoforally activeantihypertensiveagents.Science.1977;196(4288):441–4.
11. Masuyer G, Schwager SL, Sturrock ED, et al. Molecular recognitionandregulationofhumanangiotensin-I converting enzyme(ACE)activitybynaturalinhibitorypeptides.SciRep.2012;2:717.
12.SoubrierF,Alhenc-GelasF,HubertC,et al.Twoputativeactive centersinhumanangiotensinI-converting enzymerevealedby molecularcloning.ProcNatlAcadSci.1988;85(24):9386–90.
13.FuchsS, Xiao HD, HubertC, et al. Angiotensin-convertingenzymeC-terminalcatalyticdomainisthemainsiteofangio- tensinIcleavageinvivo.Hypertension.2008;51(2):267–74.
14.Ceconi C,Francolini G,OlivaresA,et al. Angiotensin-con-vertingenzyme(ACE)inhibitors havedifferentselectivityfor bradykinin bindingsitesofhumansomaticACE.EurJPhar- macol.2007;577(1–3):1–6.
15.TaddeiS,VirdisA,GhiadoniL,etal.Effectsofantihyperten-sive drugs on endothelial dysfunction: clinical implications. Drugs.2002;62(2):265–84.
16.SuwaidiJA,HamasakiS,HiganoST,etal.Long-termfollow-up ofpatientswithmildcoronaryarterydisease andendothelial dysfunction.Circulation.2000;101(9):948–54.
17.Schachinger V,BrittenMB,ZeiherAM.Prognosticimpactof coronaryvasodilatordysfunctiononadverselong-termoutcome of coronary heart disease. Circulation. 2000;101(16):1899–906.
18.Murphey LJ,HacheyDL,OatesJA,etal.Metabolism ofbra- dykinin In vivo in humans: identification of BK1-5 as a stable plasma peptide metabolite. J Pharmacol Exp Ther.2000;294(1):263–9.
19. Gauthier KM, Cepura CJ, Campbell WB. ACE inhibition enhances bradykinin relaxationsthroughnitricoxideandB1 receptor activation in bovine coronary arteries. Biol Chem.2013;394(9):1205–12.
20. BaudinB,BerardM,CarrierJL,etal.Vascularorigindeter- minesangiotensinI-converting enzymeexpressioninendothe- lialcells.Endothelium.1997;5(1):73–84.
21.CyrM,LepageY,BlaisCJr,etal.Bradykinin anddes-Arg(9)- bradykininmetabolic pathwaysandkinetics of activation of human plasma. Am J Physiol Heart Circ Physiol.2001;281(1):H275–83.
22.ErdosEG,TanF,SkidgelRA.AngiotensinI-convertingenzyme inhibitors areallostericenhancers ofkininB1andB2receptor function.Hypertension.2010;55(2):214–20.
23.Kayashima Y, Smithies O, Kakoki M. The kallikrein-kinin system and oxidative stress. Curr Opin Nephrol Hypertens.2012;21(1):92–6.
24.MaJX,WangDZ,ChaoL,et al. Cloning,sequenceanalysisand expressionofthegeneencodingthemousebradykinin B2 receptor.Gene.1994;149(2):283–8.
25.KuhrF,LowryJ,ZhangY,etal.Differential regulationof inducibleandendothelialnitricoxidesynthase bykininB1and B2receptors.Neuropeptides.2010;44(2):145–54.
26.CoutureR,GirolamiJP.Putativerolesofkininreceptors inthe therapeuticeffectsofangiotensin1-convertingenzymeinhibitors in diabetes mellitus.EurJPharmacol.2004;500(1–3):467–85.
27.Dabek J, Kulach A, Smolka G, et al. Expression of genes encoding kininreceptors inperipheralbloodmononuclearcells frompatientswithacutecoronarysyndromes.InternMedJ.2008;38(12):892–6.
28.PotierL,WaeckelL,VincentMP, etal.Selectivekininreceptor agonists ascardioprotectiveagents inmyocardialischemiaand diabetes.JPharmacolExpTher.2013;346(1):23–30.
29.DOrleans-JusteP,deNucciG,VaneJR.KininsactonB1orB2receptors toreleaseconjointlyendothelium-derivedrelaxing factorand prostacyclinfrombovineaorticendothelialcells.BrJ Pharmacol.1989;96(4):920–6.
30. WangDZ,ChaoL,ChaoJ.Hypotensionintransgenicmice overexpressinghumanbradykininB2receptor. Hypertension.1997;29(1Pt2):488–93.
31.CervenkaL,Harrison-Bernard LM,DippS,etal.Earlyonset salt-sensitive hypertension in bradykinin B(2) receptor null mice.Hypertension.1999;34(2):176–80.
32.AlfieME,Sigmon DH,PomposielloSI,etal.Effectofhighsalt intakeinmutantmicelackingbradykinin-B2 receptors.Hyper- tension.1997;29(1Pt2):483–7.
33.Ni A, Yin H, Agata J, et al. Overexpression of kinin B1 receptors induces hypertensiveresponsetodes-Arg9-bradykinin and susceptibility to inflammation. J Biol Chem.2003;278(1):219–25.
34.PesqueroJB,AraujoRC,HeppenstallPA,etal.Hypoalgesiaand alteredinflammatory responsesinmicelackingkininB1 receptors.ProcNatlAcadSci.2000;97(14):8140–5.
35.LiYY,ZhangH,XuJ,etal.Bradykinin beta2receptor-58T/C genepolymorphismandessentialhypertension:ameta-analysis. PLoSOne.2012;7(8):e43068.
36.VanGuilder GP,Pretorius M,LutherJM,etal.Bradykinintype
2receptorBE1genotypeinfluences bradykinin-dependent vasodilationduringangiotensin-converting enzymeinhibition. Hypertension.2008;51(2):454–9.
37. CuiJ,MelistaE,ChazaroI,etal.Sequencevariationofbra- dykininreceptorsB1andB2andassociationwithhypertension. JHypertens.2005;23(1):55–62.
38. DhamraitSS,PayneJR,LiP,etal.Variationinbradykinin receptorgenes increasesthecardiovascularriskassociatedwith hypertension.EurHeartJ.2003;24(18):1672–80.
39.Witherow FN, Dawson P, Ludlam CA, et al. Bradykinin
receptorantagonism andendothelialtissueplasminogen acti- vator release in humans. Arterioscler Thromb Vasc Biol.
2003;23(9):1667–70.
40.MurpheyLJ,Malave HA,PetroJ,etal.Bradykininandits metabolitebradykinin1–5inhibitthrombin-induced platelet aggregation in humans. J Pharmacol Exp Ther.
2006;318(3):1287–92.
41.Taddei S, Versari D, Cipriano A, et al. Identification of a cytochrome P450 2C9-derivedendothelium-derived hyperpo- larizing factor in essential hypertensivepatients. J Am Coll Cardiol.2006;48(3):508–15.
42.MamenkoM,ZaikaO,PochynyukO.DirectregulationofENaC bybradykinin inthedistalnephron.Implicationsforrenal sodium handling. Curr Opin Nephrol Hypertens.
2014;23(2):122–9.
43.MamenkoM,ZaikaO,DorisPA,etal.Salt-dependentinhibition ofepithelialNa?channel-mediatedsodiumreabsorption inthe aldosterone-sensitive distalnephronbybradykinin.Hyperten- sion.2012;60(5):1234–41.
44.ZaikaO,MamenkoM,ONeilRG,etal.Bradykininacutely
inhibitsactivityof theepithelialNa? channelinmammalian aldosterone-sensitive distalnephron.AmJPhysiolRenal Physiol.2011;300(5):F1105–15.
45.El-DahrSS.Spatialexpressionofthekallikrein-kininsystem
duringnephrogenesis.HistolHistopathol.2004;19(4):1301–10.
46.Bachvarov DR,LandryM,PelletierI,etal.Characterizationof twopolymorphic sitesinthehumankininB1receptorgene: alteredfrequency ofanalleleinpatientswithahistory ofend- stagerenalfailure.JAmSocNephrol.1998;9(4):598–604.
47.ZychmaMJ,GumprechtJ,Zukowska-SzczechowskaE,etal.
Polymorphismsinthegenesencodingforhumankininreceptors andtheriskofend-stage renalfailure:results oftransmission/ disequilibriumtest.TheEnd-StageRenalDiseaseStudyGroup. JAmSocNephrol.1999;10(10):2120–4.
48.JozwiakL,DropA,BuraczynskaK,etal.Associationofthe
humanbradykinin B2receptorgenewithchronicrenalfailure. MolDiagn.2004;8(3):157–61.
49. BledsoeG,ShenB,YaoY,etal.Reversalofrenalfibrosis, inflammation,andglomerularhypertrophy bykallikreingene delivery.HumGeneTher.2006;17(5):545–55.
50.KakokiM,McGarrahRW,KimHS,etal.BradykininB1andB2 receptors bothhaveprotectiverolesinrenalischemia/reperfu- sioninjury.ProcNatlAcadSci.2007;104(18):7576–81.
51.HarrisonDG,GongoraMC.Oxidativestressandhypertension.
MedClinNAm.2009;93(3):621–35.
52.SangsreeS,Brovkovych V,MinshallRD,etal.KininaseI-type carboxypeptidases enhancenitricoxideproductioninendothe- lialcellsbygeneratingbradykinin B1receptoragonists.AmJ PhysiolHeartCircPhysiol.2003;284(6):H1959–68.
53.GryglewskiRJ,UraczW,ChlopickiS,etal.Bradykininasa
major endogenous regulator of endothelial function. Pediatr
PatholMolMed.2002;21(3):279–90.
54.OeseburgH,IusufD,vanderHarst P,etal.Bradykininprotects againstoxidativestress-induced endothelialcellsenescence. Hypertension.2009;53(2):417–22.
55. DelemasureS,BlaesN,RichardC,etal.Antioxidant/oxidant statusandcardiac functioninbradykininB(1)-andB(2)-re- ceptornullmice.Physiol ResAcademiaScientiarumBohe- moslovaca.2013;62(5):511–7.
56.BentleyJP,AsselbergsFW,CoffeyCS,etal.Cardiovascular riskassociatedwithinteractionsamongpolymorphismsingenes fromtherenin-angiotensin,bradykinin,and fibrinolyticsystems. PLoSOne.2010;5(9):e12757.
57.Thogersen AM,JanssonJH,BomanK,etal.Highplasminogen activatorinhibitorandtissueplasminogen activatorlevelsin plasmaprecedeafirst acutemyocardialinfarctioninbothmen andwomen:evidenceforthefibrinolytic systemasaninde- pendentprimaryriskfactor.Circulation.1998;98(21):2241–7.
58.WimanB,AnderssonT,HallqvistJ,etal.Plasmalevelsof
tissueplasminogen activator/plasminogenactivatorinhibitor-1 complexandvonWillebrandfactoraresignificantriskmarkers forrecurrentmyocardialinfarctionintheStockholm Heart Epidemiology Program(SHEEP)study. ArteriosclerThromb VascBiol.2000;20(8):2019–23.
59.AsselbergsFW,WilliamsSM,HebertPR,etal.Epistaticeffects ofpolymorphismsingenesfromtherenin-angiotensin, brady- kinin,andfibrinolyticsystemsonplasmat-PAand PAI-1levels. Genomics.2007;89(3):362–9.
60.LabinjohC,NewbyDE,DawsonP,etal.Fibrinolyticactionsof intra-arterialangiotensin IIandbradykinin invivoinman. CardiovascRes.2000;47(4):707–14.
61.Rahman AM, Murrow JR, Ozkor MA, et al. Endothelium- derivedhyperpolarizingfactor mediatesbradykinin-stimulated tissueplasminogenactivatorrelease inhumans.JVascRes.
2014;51(3):200–8.
62. TaddeiS,VirdisA,GhiadoniL,etal.VitaminCimproves endothelium-dependent vasodilation by restoringnitric oxide activity in essential hypertension. Circulation.
1998;97(22):2222–9.
63.Giannarelli C, Virdis A, De Negri F, et al. Effect of sul- faphenazoleontissueplasminogen activatorreleaseinnor- motensive subjects and hypertensive patients. Circulation.
2009;119(12):1625–33.
64. BrownNJ,NadeauJH,VaughanDE.Selectivestimulationof tissue-typeplasminogen activator(t-PA)invivobyinfusionof bradykinin.ThrombHaemost.1997;77(3):522–5.
65.KimuraS,TsujiH,NishimuraH,etal.Bradykininenhances invitroprocoagulantandantifibrinolytic propertiesofratvas- cularendothelialcells.ThrombRes.2002;106(1):41–50.
66.RegoliD,GobeilFJr.Criticalinsightsintothebeneficial and protectiveactionsofthekallikrein-kininsystem. Vascul Phar- macol.2015;64:1–10.
67.YehCH,Chen TP,Wang YC,etal.Cardiomyocyticapoptosis limitedbybradykinin viarestoration ofnitricoxideaftercar- dioplegicarrest.JSurgRes.2010;163(1):e1–9.
68. ChaoJ,LiHJ,YaoYY,etal.Kinininfusionpreventsrenal inflammation,apoptosis,andfibrosisviainhibitionofoxidative stressandmitogen-activatedproteinkinase activity.Hyperten- sion.2007;49(3):490–7.
69. PerezV,VelardeV,Acuna-CastilloC,etal.Increasedkinin levels and decreased responsivenessto kinins during aging. JGerontolABiolSciMedSci.2005;60(8):984–90.
70.Kuoppala A,ShiotaN,LindstedtKA,etal.Expression ofbra- dykininreceptorsin the left ventricles ofratswithpressure overload hypertrophy and heart failure. J Hypertens.
2003;21(9):1729–36.
71.NurmiL,HeikkilaHM,VapaataloH,etal.Downregulation of Bradykinintype2receptor expressionincardiac endothelial cellsduringsenescence.JVascRes.2012;49(1):13–23.
72.LiesmaaI,ShiotaN,KokkonenJO,etal.Bradykinintype-2
receptor expression correlates with age and is subjected to transcriptionalregulation.IntJVascMed.2012;2012:159646.
73.Gerhard M,Roddy MA,CreagerSJ, etal.Aging progressively impairsendothelium-dependentvasodilationin forearmresis- tancevesselsofhumans.Hypertension.1996;27(4):849–53.
74.Taddei S, Virdis A, Mattei P, et al. Aging and endothelial functioninnormotensive subjectsandpatientswithessential hypertension.Circulation.1995;91(7):1981–7.
75.Sharma JN. Hypertension and the bradykinin system. Curr
HypertensRep.2009;11(3):178–81.
76.Ceconi C, Fox KM, Remme WJ, et al. ACE inhibition with perindoprilandendothelialfunction.Resultsofasubstudy ofthe EUROPAstudy:PERTINENT.CardiovascRes.2007;73(1):237–46.
77.Campbell DJ, KladisA, DuncanAM.Effects of converting enzymeinhibitorsonangiotensinandbradykinin peptides. Hypertension.1994;23(4):439–49.
78.Bujak-Gizycka B,OlszaneckiR,MadejJ,etal.Metabolismof bradykininin aorta of hypertensiverats. Acta Biochim Pol.
2011;58(2):199–202.
79.SuJB,BarbeF,CrozatierB,etal.Increasedbradykinin levels accompanythehemodynamicresponse toacuteinhibitionof angiotensin-converting enzyme in dogs with heart failure. JCardiovascPharmacol.1999;34(5):700–10.
80.Gainer JV,Morrow JD,LovelandA,etal.Effectofbradykinin- receptorblockadeontheresponse toangiotensin-converting- enzyme inhibitor in normotensiveandhypertensivesubjects. NEnglJMed.1998;339(18):1285–92.
81.SquireIB,OKaneKP,AndersonN,etal. BradykininB(2) receptorantagonismattenuatesbloodpressure responsetoacute angiotensin-converting enzymeinhibitioninnormalmen. Hypertension.2000;36(1):132–6.
82.TaddeiS,VirdisA,GhiadoniL,etal.Effectsofangiotensin
converting enzyme inhibition on endothelium-dependent vasodilatationinessentialhypertensivepatients.JHypertens.
1998;16(4):447–56.
83.Witherow FN,DawsonP,LudlamCA,etal.Markedbradyki- nin-inducedtissueplasminogenactivator release in patients with heartfailuremaintainedon long-termangiotensin-converting enzyme inhibitor therapy.JAmCollCardiol.2002;40(5):961–6.
84.Lang NN, Cruden NL, Tse GH, et al. Vascular B1 kinin
receptorsinpatientswithcongestiveheartfailure.JCardiovasc
Pharmacol.2008;52(5):438–44.
85.BrugtsJJ, NinomiyaT,BoersmaE,etal.Theconsistencyofthe treatmenteffectofanACE-inhibitorbased treatmentregimenin patientswithvascular diseaseorhighriskofvasculardisease:a combinedanalysisofindividualdataofADVANCE,EUROPA, andPROGRESStrials.EurHeartJ.2009;30(11):1385–94.
86. LabinjohC,NewbyDE,PellegriniMP,etal.Potentiationof bradykinin-induced tissueplasminogenactivatorreleaseby angiotensin-convertingenzymeinhibition.JAmCollCardiol.
2001;38(5):1402–8.
87.FogariR,MugelliniA,ZoppiA,etal.Losartan andperindopril effects on plasma plasminogenactivator inhibitor-1and fib- rinogeninhypertensive type2diabeticpatients.AmJHyper- tens.2002;15(4Pt1):316–20.
88. GohlkeP,LambertyV,KuwerI,etal.Long-termlow-dose angiotensin convertingenzymeinhibitortreatmentincreases vascularcyclicguanosine30,50-monophosphate.Hypertension.
1993;22(5):682–7.
89.RumbleJR,Komers R,Cooper ME.Kininsornitricoxide,or both,areinvolved intheantitrophiceffectsofangiotensin converting enzymeinhibitorsondiabetes-associatedmesenteric vascularhypertrophyintherat.JHypertens.1996;14(5):601–7.
90.BuleonM,AllardJ,JaafarA,etal.Pharmacological blockadeof B2-kininreceptorreduces renalprotectiveeffectofangiotensin- converting enzyme inhibition in db/db mice model. Am J PhysiolRenalPhysiol.2008;294(5):F1249–56.
91.BrugtsJJ,IsaacsA,Boersma E,etal.Geneticdeterminantsof treatmentbenefitoftheangiotensin-convertingenzyme-inhibitor perindopril inpatientswithstablecoronaryarterydisease.Eur HeartJ.2010;31(15):1854–64.
92.GaoL,YuDM.Molecularmechanismoflimbs postischemic revascularizationimprovedbyperindopril indiabeticrats.Chin MedJ(Engl).2008;121(21):2129–33.
93.IgnjatovicT,StanisavljevicS,Brovkovych V,etal.KininB1 receptorsstimulatenitricoxideproduction inendothelialcells: signaling pathways activated by angiotensin I-converting enzyme inhibitors and peptide ligands. Mol Pharmacol.
2004;66(5):1310–6.
94. NgK,ButlinM,AvolioAP.Persistenteffectofearly,brief angiotensin-convertingenzyme inhibition onsegmentalpressure dependencyofaortic stiffnessin spontaneouslyhypertensive rats.JHypertens.2012;30(9):1782–90.
95.OndettiMA.Structuralrelationshipsofangiotensinconverting-
enzyme inhibitors to pharmacologic activity. Circulation.
1988;77(6Pt2):I74–8.
96.FerrariR.Preservingbradykinin orblockingangiotensin II:the cardiovascular dilemma. Dialogues Cardiovasc Med.
2004;9(20):71–89.
97.CeconiC,FrancoliniG,BastianonD,etal.Differences inthe effectofangiotensin-convertingenzymeinhibitorsonthe rateof endothelialcellapoptosis: invitroandinvivostudies.Cardio- vascDrugsTher.2007;21(6):423–9.
98.Taddei S.RASinhibitorsdose-dependentefficacy:myth or
reality?CurrMedResOpin.2015;31(7):1245–56.
99. TsoukasG,AnandS,YangK,etal.Dose-dependentantihy- pertensive efficacyandtolerability ofperindoprilinalarge, observational, 12-week,generalpractice-basedstudy.AmJ CardiovascDrugs.2011;11(1):45–55.
100. StojanovV,OtasevicP,TasicN,etal.Perindoprilantihyper- tensiveefficacy ingrade1andgrade2hypertensivepatients [PosterPP.05.131].JHypertens.2013;SupplA:e181.
101.MakaniH,BangaloreS,Supariwala A,etal.Antihypertensive efficacyof angiotensinreceptor blockersas monotherapyas evaluatedbyambulatorybloodpressure monitoring:ameta- analysis.EurHeartJ.2014;35(26):1732–42.
102.Elgendy IY, Huo T, Chik V, et al. Efficacyand safety of angiotensinreceptorblockersinolderpatients:ameta-analysis ofrandomizedtrials.AmJHypertens.2015;28(5):576–85.
103. BussienJP,dAmoreTF,PerretL,etal.Singleandrepeated dosing oftheconvertingenzymeinhibitorperindopriltonormal subjects.ClinPharmacolTher.1986;39(5):554–8.
104.Johnston C. Effects of three single doses of perindoprilon
bradykinin,kallikreinandtherenin-angiotensinsystem: double- blindplacebocontrolledstudy in12healthyvolunteers.1994. ServierLaboratories,France.
105.TropeanoAI,BoutouyrieP,PannierB,etal.Brachialpressure-
independentreductionincarotidstiffness afterlong-term angiotensin-converting enzymeinhibitionindiabetichyperten- sives.Hypertension.2006;48(1):80–6.
106.RemmeWJ,DeckersJW,FoxKM,etal.Secondary prevention ofcoronarydiseasewithACEinhibition–does bloodpressure reductionwithperindoprilexplainthebenefits inEUROPA? CardiovascDrugsTher.2009;23(2):161–70.
107.Lonn E, Yusuf S, DzavikV, et al. Effects of ramipril and vitaminEonatherosclerosis: thestudytoevaluatecarotid ultrasound changesinpatientstreatedwithramiprilandvitamin E(SECURE).Circulation.2001;103(7):919–25.
108. YusufS,SleightP,PogueJ,etal.Effectsofanangiotensin- converting-enzymeinhibitor,ramipril,on cardiovascularevents inhigh-riskpatients.TheHeartOutcomes PreventionEvalua- tionStudyInvestigators.NEnglJMed.2000;342(3):145–53.
109.FoxKM.Efficacy ofperindoprilinreductionofcardiovascular eventsamongpatientswithstablecoronary arterydisease:ran- domised,double-blind,placebo-controlled,multicentretrial(the EUROPAstudy).Lancet.2003;362(9386):782–8.
110.Bangalore S, Kumar S, Messerli FH. Angiotensin-converting enzymeinhibitorassociatedcough:deceptiveinformationfromthe PhysiciansDeskReference.AmJMed.2010;123(11):1016–30.
111. BritishHypertensionSociety.Angiotensinconvertingenzyme (ACE)inhibitors 2008[citedNovember 21,2014];Available from: http://www.bhsoc.org/pdfs/therapeutics/Angiotensin%20
Converting%20Enzyme%20(ACE)%20Inhibitors.pdf.
112.MukaeS,ItohS, AokiS, etal.Associationof polymorphismsof therenin-angiotensin systemandbradykininB2receptorwith ACE-inhibitor-related cough. J Hum Hypertens.
2002;16(12):857–63.
113.MaliniPL,StrocchiE,ZanardiM,etal.Thromboxane antago- nismandcough inducedbyangiotensin-converting-enzyme inhibitor.Lancet.1997;350(9070):15–8.


{kind=link}