

ThS. BS TRẦN CÔNG DUY1
NGUYỄN CHÂU TUẤN2
1 Giảng viên bộ môn Nội Tổng quát, Đại học Y Dược TPHCM
2 Bác sĩ nội trú bộ môn Lão Khoa, Đại học Y Dược TPHCM
ĐẠI CƯƠNG
Bệnh cơ tim phì đại (BCTPĐ) là tình trạng tăng độ dày thành tim (≥ 15mm ở người trưởng thành, hoặc ≥ 13mm ở người trưởng thành có người thân trực hệ đời thứ nhất bị BCTPĐ) ở một hoặc nhiều hơn các vùng của thành thất trái, mà không thể giải thích được bằng các tình trạng bất thường về tải áp lực [14], [15].
Hầu hết các bệnh nhân có phì đại vách liên thất không đối xứng và khoảng 40 – 70% có BCTPĐ tắc nghẽn, được chẩn đoán bởi chênh áp trong buồng thất trái ≥ 30 mmHg lúc nghỉ (khoảng 25% bệnh nhân) hoặc lúc gắng sức [15], [39],[52]. BCTPĐ không tắc nghẽn, có chênh áp < 30mmHg lúc nghỉ và/hoặc lúc gắng sức, hiện diện ở 30 – 60% bệnh nhân [15], [39], [52].
Trong một số báo cáo ở Châu Âu, Châu Á và Bắc Mỹ, tỉ lệ hiện mắc của BCTPĐ là 2-5 ca trên 1000 dân [9],[22], [47],[78]. Trong 60% bệnh nhân, BCTPĐ là hậu quả của những đột biến gen sarcomere theo nhiễm sắc thể trội, trong khi những nguyên nhân khác bao gồm các hội chứng di truyền, các rối loạn thần kinh cơ và các bệnh lý dự trữ đặc trưng bởi sự tích tụ các chất bất thường trong gian bào (như bệnh Anderson-Fabry, Pompe, hoặc Danon, v.v…) chiếm từ 5 – 10% bệnh nhân[14], [44]. Ở khoảng 30% bệnh nhân, nguyên nhân của BCTPĐ vẫn chưa rõ [15]. Đánh giá các nguyên nhân của suy tim trong BCTPĐ được trình bày trong hình 1.
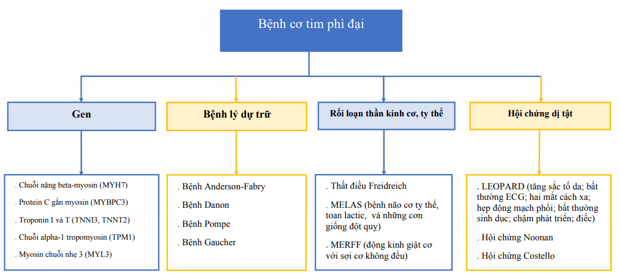
Hình 1. Các nguyên nhân của BCTPĐ
Suy tim có hai đặc điểm lâm sàng khác biệt trong BCTPĐ; trong phần lớn các bệnh nhân, suy tim có biểu hiện kiểu hình là suy tim phân suất tống máu bảo tồn (HFpEF), với đặc điểm đặc biệt ở các bệnh nhân là tắc nghẽn đường ra thất trái, trong khi chỉ có một phần nhỏ bệnh nhân tiến triển suy tim phân suất tống máu giảm ở giai đoạn sau. Do sự không đồng nhất về nguyên nhân và lâm sàng, việc xác định tỉ lệ mới mắc của suy tim do BCTPĐ là một thách thức. Dữ liệu từ một nghiên cứu đoàn hệ với 1000 bệnh nhân được chẩn đoán BCTPĐ ở tuổi trung niên (30 – 59 tuổi) cho thấy tỉ lệ mới mắc của suy tim khoảng 50%, với triệu chứng thay đổi từ nhẹ đến nặng (NYHA II – IV) [34]. Trong nghiên cứu sổ bộ trên 3208 người có bệnh cơ tim ở Châu Âu, tỉ lệ hiện mắc của suy tim có triệu chứng ở những bệnh nhân có BCTPĐ là 67% (triệu chứng NYHA II và III – IV lần lượt là 49,9% và 17,4%) [8]. Tuy nhiên, tỉ lệ hiện mắc được đề cập của suy tim do BCTPĐ có thể bị ước tính quá mức do sự ít đại diện của các đối tượng có triệu chứng nhẹ hoặc không có triệu chứng trong hệ thống dữ liệu. Suy tim thường gặp trong phần lớn các bệnh nhân có BCTPĐ tắc nghẽn và 10% bệnh nhân với BCTPĐ không tắc nghẽn [35]. Suy tim cấp không thường gặp, tuy nhiên có thể bị thúc đẩy bởi các tình trạng như loạn nhịp nhanh (như rung nhĩ), thiếu máu cục bộ cơ tim, hở van hai lá cấp hoặc nặng lên (như đứt dây thừng gân), hoặc bệnh đồng mắc (như nhiễm độc giáp) [15], [27],[49]. Tiến triển đến suy tim nặng (như triệu chứng NYHA II – IV) xảy ra ở 3,5 – 17% bệnh nhân, thường là hậu quả của tắc nghẽn đường ra thất trái, hoặc do tái cấu trúc thất trái làm rối loạn chức năng tâm thu [23], [43], [50], [71]. Tỉ lệ hiện mắc của suy tim do BCTPĐ không có sự khác biệt về giới hoặc chủng tộc, nhưng những bệnh nhân có bệnh protein sarcomere có xu hướng tiến triển thành suy tim ở tuổi trẻ hơn và có xu hướng suy tim tiến triển cao hơn so với những bệnh nhân không có các đột biến [51]. Những bệnh nhân có các rối loạn di truyền hiếm (như bệnh Anderson-Fabry, Danon hoặc bệnh ty thể) thường có bệnh lý đa hệ thống, nhưng biểu hiện lâm sàng thường (khoảng 60%) nổi bật với các triệu chứng của suy tim cũng như các bất thường về dẫn truyền[42].
Ở những bệnh nhân suy tim, những người có BCTPĐ chiếm 2 – 3% [72]. Tỉ lệ bệnh nhân BCTPĐ được ghép tim vì suy tim nặng hơi thấp hơn so với những nguyên nhân khác, vì BCTPĐ là một bệnh hiếm [77]. Tuy nhiên, khi so với những bệnh nhân khác được ghép tim, những bệnh nhân BCTPĐ thường trẻ hơn và có ít bệnh đồng mắc hơn tại cùng thời điểm ghép tim. Điều này cũng làm cho tiên lượng ngắn hạn và dài hạn sau ghép tương đương hoặc khả quan hơn [77].
SINH LÝ BỆNH
Bệnh cơ tim phì đại do đột biến gen sarcomere
Ở bệnh nhân BCTPĐ do gen, phì đại và xáo trộn tế bào cơ tim xảy ra để đáp ứng với rối loạn cân bằng năng lượng do tiêu thụ năng lượng quá mức nhằm tạo ra sức căng trong sarcomere[17], [70]. Mất cân bằng năng lượng, cùng với nhu cầu oxy cao của cơ tim phì đại dẫn đến các đợt thiếu máu cục bộ cơ tim do tăng nhu cầu (như khi gắng sức hoặc tim nhanh) có thể giải thích các triệu chứng đau ngực, giảm khả năng gắng sức và khó thở khi gắng sức [31]. Ở một vài bệnh nhân, cơ chế sinh lý bệnh có thể nặng nề hơn do quá tải huyết động gây tắc nghẽn động đường ra thất trái, ở giữa buồng hoặc đa tầng[32]. Rối loạn chức năng vi mạch vành, đặc trưng bởi bất thường cấu trúc và giảm lưu lượng dòng máu trong những động mạch vành trong thành tim, cũng đóng một vai trò quan trọng trong các đợt thiếu máu cơ tim tái phát và hình thành suy tim [38]. Ngoài ra, rối loạn kết thúc co cơ với nồng độ Ca2+ nội bào thấp làm cơ dãn ra không hoàn toàn và rối loạn chức năng tâm trương có thể xuất hiện trước và sau sự phát triển phì đại quá mức [24].
Ở một số bệnh nhân, sự tích luỹ ảnh hưởng của các yếu tố này làm thiếu hụt năng lượng trong tế bào cơ tim sau đó dẫn đến việc mất tế bào cơ tiến triển và thay thế bởi mô xơ, cuối cùng dẫn đến tái cấu trúc thất trái và rối loạn chức năng tâm thu và suy tim phân suất tống máu giảm. Thật vậy, trong một phân tích gộp gồm 1063 bệnh nhân BCTPĐ được theo dõi trung bình 3,1 năm cho thấy sự thay thế bởi mô xơ trên cộng hưởng từ bắt tính hiệu cản từ muộn LGE-CMR tiên đoán tăng nguy cơ tử vong có ý nghĩa do suy tim [21]. Trong một nghiên cứu về mô học trên 30 trái tim ghép ở giai đoạn cuối của BCTPĐ, hơn 1/3 cơ tim thất trái bị thay thế bởi mô xơ, đặc biệt ở vùng mỏm thất trái và vách liên thất [18]. Những bệnh nhân có đột biến đa gen trong protein sarcomere (chiếm đến 5% dân số BCTPĐ) có nhiều nguy cơ thúc đẩy tiến triển đến bệnh giai đoạn cuối [19],[20]. Ngoài ra, yếu tố gia đình có suy tim nặng được ghi nhận là một chỉ dấu nguy cơ của kết cục xấu ở các thành viên khác trong gia đình [23]. Bên cạnh đó, các bệnh đồng mắc (như viêm cơ tim hoặc bệnh động mạch vành thượng tâm mạc) có thể liên quan đến tái cấu trúc thất trái và tiến triển thành suy tim toàn phát [52].
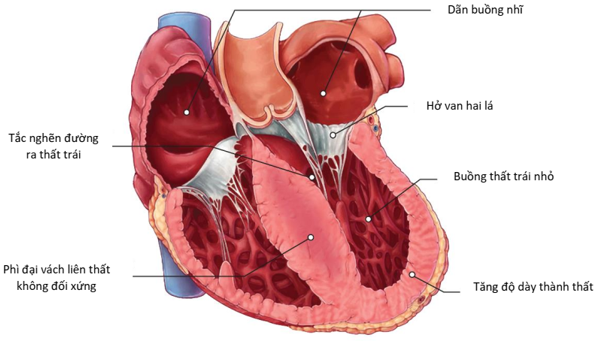
Ở những bệnh nhân BCTPĐ tắc nghẽn, độ nặng của suy tim được quyết định bởi quá tải áp lực gây ra bởi tắc nghẽn động học đường ra thất trái trong thời kì tâm thu [35]. Những thay đổi hình thái đặc trưng liên quan suy tim trong BCTPĐ được tóm tắt trong hình 2. Tắc nghẽn trong buồng tim thường gặp nhất liên quan đến tắc nghẽn đường ra và gây ra bởi kết hợp tắc nghẽn vật lý của phì đại mô vách liên thất, cử động ra trước bất thường trong kì tâm thu của van hai lá (SAM) và những khiếm khuyết co và dãn cơ tim. Ở 5 – 10% bệnh nhân, chênh lệch áp lực gây ra duy nhất bởi tắc nghẽn giữa buồng tim do vị trí bất thường của vùng vách liên thất phì đại và cơ nhú trước bên [35]. Các thay đổi động học trong chênh áp để đáp ứng với các thay đổi về co bóp cơ tim và tình trạng tải (như gắng sức, dịch) giải thích sự thay đổi theo thời gian và xuất hiện chậm của các triệu chứng của suy tim trong BCTPĐ [35]. Ở những bệnh nhân không có chênh áp nhiều lúc nghỉ, các nghiệm pháp gắng sức tim phổi là biện pháp được ưa thích sử dụng để phát hiện tắc nghẽn [14]. Rối loạn chức năng tâm trương thất trái đại diện cho một cơ chế quan trọng khác trong việc hình thành suy tim (như suy tim với phân suất tống máu thất trái bảo tồn) trong BCTPĐ. Rối loạn chức năng tâm trương xuất hiện ở hầu hết các bệnh nhân, bất kể có tắc nghẽn buồng tim hay không và được đặc trưng bởi dãn đẳng trường kéo dài và giảm khả năng đổ đầy thất [52]. Bên cạnh đó, bất thường van hai lá, cầu cơ mạch vành, phình mỏm tim, tái cấu trúc nhĩ và rối loạn chức năng tự động có thể góp phần vào việc hình thành và độ nặng của suy tim [4], [36], [46],[50], [64], [67],[69]. Ở những bệnh nhân BCTPĐ không tắc nghẽn, suy tim phần lớn là do rối loạn chức năng tâm trương, nhưng ở một số bệnh nhân, suy tim có thể là tiến triển của giai đoạn cuối của bệnh và suy tim với phân suất tống máu thất trái giảm nặng.
Hình 2. Các thay đổi hình thái của tim trong BCTPĐ
Bệnh cơ tim phì đại do rối loạn dự trữ
Ở những bệnh nhân BCTPĐ do rối loạn dự trữ hiếm gặp (như bệnh Anderson-Fabry, Danon và Pompe), suy tim hầu hết có kiểu hình suy tim với phân suất tống máu bảo tồn do dày thành thất trái đồng tâm, lan toả [1],[76]. Tăng độ dày thành tim gây ra một phần do phì đại cơ tim (do tích tụ trong tiêu thể glycosphingolipids), và một phần do xơ hoá mô kẽ bị kích hoạt bởi sự sản xuất quá mức của các cytokines gây xơ hoá [1]. Phì đại không đồng tâm trong các rối loạn dự trữ hiếm gặp (< 2,5%), trong khi phì đại hai thất có thể xảy ra ở 25% bệnh nhân [76]. Trong bệnh Anderson-Fabry, mô xơ (phát hiện bằng hình ảnh bắt cản từ muộn trên MRI tim hoặc bằng các hình ảnh biến đổi) trong thành sau bên có thể góp phần gây rối loạn chức năng thất trái và hở hai lá cơ năng [75]. Hầu hết bệnh nhân có chức năng thất trái bảo tồn, thỉnh thoảng có bằng chứng của tắc nghẽn thất trái dưới van động mạch chủ [5]. Suy tim toàn phát được quyết định bởi mức độ rối loạn chức năng tâm trương thất trái, tương quan với phạm vi phì đại thất trái và nồng độ N-terminal pro B-type natriuretic peptide (NT-proBNP) [74]. Hiếm khi, rối loạn chức năng tâm trương trong các rối loạn dự trữ có thể tiến triển đến kiểu hạn chế đổ đầy, đi kèm với dãn rộng buồng nhĩ hai bên [76]. Ở những bệnh nhân dó, tổn thương tim có thể có đặc điểm của bệnh cơ tim hạn chế; do đó, các rối loạn dự trữ cần được xem xét là nguyên nhân của cả BCTPĐ và bệnh cơ tim hạn chế. Sự hình thành rối loạn chức năng tâm thu và suy tim phân suất tống máu giảm luôn luôn xảy ra trong bệnh Danon và thường ở những bệnh nhân có các bệnh cơ tim do chuyển hoá khác [33].
DIỄN BIẾN TỰ NHIÊN VÀ KẾT CỤC
Bệnh cơ tim phì đại do đột biến gen sarcomere
Phì đại thất trái điển hình trong BCTPĐ gây ra do các rối loạn sarcomere thường xuất hiện ở tuổi thiếu niên hoặc trưởng thành (mặc dù có thể xuất hiện ở thời thơ ấu đến khoảng 70 tuổi), và vẫn ổn định với chức năng tâm thu thất trái bảo tồn và rối loạn chức năng tâm trương với nhiều mức độ [53]. Ở những bệnh nhân có BCTPĐ tắc nghẽn, độ nặng và tiên lượng của suy tim được quyết định chủ yếu bởi tắc nghẽn đường ra thất trái. Điều này được làm rõ bởi các dữ liệu cho thấy chênh áp ≥ 30 mmHg lúc nghỉ tiên lượng độc lập với suy tim tiến triển và tăng tỉ lệ tử vong [37]. Các dữ kiện gần đây từ một nghiên cứu đoàn hệ với 324 bệnh nhân có BCTPĐ tắc nghẽn và suy tim nhẹ ban đầu chứng minh sự tiến triển đến phân độ chức năng NYHA III – IV, với tỉ lệ hàng năm 3,2 – 7,4% phụ thuộc vào mức độ tắc nghẽn đường ra thất trái [40]. Suy tim nặng xuất hiện ở 20 – 38% bệnh nhân với thời gian theo dõi 6,5 năm [40]. Tương tự, trong một nghiên cứu đoàn hệ gồm 293 bệnh nhân BCTPĐ với thời gian theo dõi trung bình 6 năm, suy tim tiến triển xuất hiện ở 20% bệnh nhân có tắc nghẽn đường ra thất trái nặng [43]. Các đặc điểm phân biệt ở những bệnh nhân này là tuổi cao (50 ± 14 tuổi) và tăng có ý nghĩa độ dày thành thất trái [43].
Tắc nghẽn giữa buồng tim thường đi kèm với triệu chứng suy tim nặng và giảm tỉ lệ sống còn. Trong một nghiên cứu đoàn hệ của 423 bệnh nhân, tắc nghẽn giữa buồng tim được tìm thấy ở 8% bệnh nhân, những bệnh nhân này có nhiều triệu chứng (> 90% với NYHA ≥ II) và có tỉ lệ tử vong cao hơn so với những đối tượng còn lại [12].
Rối loạn chức năng tâm trương nặng (giảm đổ đầy) có thể thấy ở 9,2% bệnh nhân có BCTPĐ, thường ở bệnh cảnh phì đại cơ tim nặng, kèm hoặc không kèm tắc nghẽn đường ra thất trái. Những bệnh nhân này thường có các triệu chứng của cung lượng tim thấp (hơn là sung huyết), và thường có tăng nguy cơ độc lập tiến triển đến suy tim nặng và bệnh giai đoạn cuối [3],[43]. Trong một nghiên cứu, những bệnh nhân này chiếm 48% các trường hợp suy tim tiến triển, và là hậu quả của giảm đổ đầy, họ có dãn nhĩ trái tại thời điểm nhận vào nghiên cứu hoặc trong quá trình theo dõi.
Ở những bệnh nhân BCTPĐ không tắc nghẽn, bệnh thường diễn tiến ổn định, lành tính và phần lớn vẫn không có suy tim hoặc chỉ có triệu chứng nhẹ do rối loạn chức năng tâm trương. Tuy nhiên, trong 7 – 10% bệnh nhân có BCTPĐ không tắc nghẽn (tỉ lệ mới mắc 1,6% mỗi năm) [35], [55], bệnh có thể tiến triển với đặc điểm dãn lớn thất trái, mỏng thành tim, và tiến triển rối loạn chức năng tâm thu thất trái, gồm có phân suất tống máu thất trái trong giới hạn bình thường – thấp [53],[63]. Tái cấu trúc thất trái bất lợi phụ thuộc bởi lượng cơ tim bị thay thế bởi mô xơ [53],[48]. Giai đoạn nặng nề nhất xảy ra ở 3% bệnh nhân và có nguy cơ tử vong đáng kể (11% mỗi năm)[23].
Ngoài ra, dãn lớn nhĩ trái và phải được công nhận là yếu tố tiên đoán độc lập của kết cục xấu của BCTPĐ [28],[40],[43]. Tương tự, tần suất của rung nhĩ, thường ở tuổi trẻ hơn dân số chung, làm tăng đáng kể nguy cơ của diễn tiến lâm sàng bất lợi [43],[49].
Bệnh cơ tim phì đại do rối loạn dự trữ
Ở những bệnh nhân BCTPĐ do các rối loạn dự trữ di truyền, suy tim có thể biểu hiện rõ ràng ở bất kì thời điểm nào từ lúc trẻ đến lúc trưởng thành tuỳ thuộc vào mức độ tổn thương tim và liên quan với độ nặng của thiếu hụt enzyme [30]. Trong bệnh Anderson-Fabry, sự xuất hiện của suy tim toàn phát được ghi nhận trong 23% bệnh nhân, thường thấy ở khoảng 30 đến 50 tuổi [29]. Tiến triển đến suy tim nặng thường gặp ở 10% bệnh nhân qua theo dõi trung bình 7,1 năm [56]. Tăng nồng độ các dấu ấn sinh học tim (troponin T, NT-proBNP) và xơ hoá nặng có liên quan với giảm chức năng tâm thu thất trái trong thời gian theo dõi [65]. Bệnh lý tim có thể tiến triển đến rối loạn chức năng tâm thu thất trái và suy tim với phân suất tống máu thất trái giảm trong 6 – 8% (đặc biệt không có điều trị thay thế enzyme) và có nguy cơ tử vong cao liên quan đến suy tim [62], [66].
ĐIỀU TRỊ
Điều trị suy tim ở những bệnh nhân BCTPĐ bao gồm biện pháp điều trị chung cho suy tim và điều trị nguyên nhân.
Điều trị hàng đầu ở những bệnh nhân BCTPĐ bao gồm ức chế beta không dãn mạch để làm giảm co bóp và làm giảm những hậu quả của rối loạn chức năng tâm trương thất trái bằng cách giảm nhịp tim, phối hợp với lợi tiểu quai liều thấp để kiểm soát các triệu chứng của suy tim, trong khi tránh giảm thể tích. Ở những bệnh nhân không dung nạp hoặc có chống chỉ định với ức chế beta, verapamil hoặc diltiazem có thể được thay thế. Tuy nhiên, y văn vẫn còn ít các chứng cứ cho thấy cách những thuốc này ảnh hưởng lên diễn tiến tự nhiên và kết cục của BCTPĐ [15].
Ở những bệnh nhân BCTPĐ tắc nghẽn và phân suất tống máu thất trái bảo tồn mà vẫn còn triệu chứng dù đã dùng liều tối đa của ức chế beta, disopyramide có thể được cân nhắc là thuốc hàng thứ hai, thêm vào điều trị [15]. Disopyramide có hoạt tính inotrope âm có thể làm giảm chênh áp dòng chảy đường ra thất trái ở phần lớn bệnh nhân và cải thiện những triệu chứng suy tim, mà không ảnh hưởng lên tử vong, hoặc thúc đẩy loạn nhịp tim [68]. Ở những bệnh nhân BCTPĐ tắc nghẽn có chênh áp ≥ 50 mmHg lúc nghỉ, hoặc khi gắng sức, và vẫn còn triệu chứng (NYHA III-IV) dù đã điều trị theo khuyến cáo, biện pháp làm giảm chênh áp xâm lấn như phẫu thuật cắt vách liên thất hoặc chích alcohol vào thành thất nên được cân nhắc [15]. Phẫu thuật cắt vách liên thất được chứng minh làm giảm đáng kể hoặc hoàn toàn tắc nghẽn > 90% bệnh nhân được điều trị ở những trung tâm có kinh nghiệm, sau đó cũng cải thiện triệu chứng suy tim dài hạn và cải thiện tỉ lệ sống còn [54], [60]. Một cách khác, chích alcohol vách liên thất được cho thấy mang lại sự cải thiện kết cục so với phẫu thuật [26]. Phẫu thuật dường như ít có lợi ở những bệnh nhân lớn tuổi hoặc những người có rung nhĩ dai dẳng [11]. Chích alcohol vách liên thất có thể ít hiệu quả ở những bệnh nhân trẻ tuổi với mức chênh áp ban đầu cao [16]. Biến chứng quanh thủ thuật của cả hai thủ thuật bao gồm có block nhĩ thất (7 – 20%), block nhánh, hoặc thủng vách liên thất [15]. Những phương pháp điều trị này hiện có ở một số ít trung tâm có kinh nghiệm.
Đặt máy tạo nhịp hai buồng không chứng minh được lợi ích trong điều trị [59]. Hiện tại, phương pháp này được khuyến cáo ở những bệnh nhân có BCTPĐ tắc nghẽn (và có chỉ định tạo nhịp chống nhịp chậm), mà không thích hợp hoặc không có nguyện vọng phẫu thuật hoặc chích alcohol [15].
Những bệnh nhân BCTPĐ tăng nguy cơ của đột tử do tim. Để phòng ngừa nguyên phát, hướng dẫn của Hội Tim Châu Âu khuyến cáo sử dụng một mô hình tiên đoán đã được chuẩn hoá (như HCM Risk-SCD) để ước tính nguy cơ đột tử do tim 5 năm [15]. Đặc biệt, ở những bệnh nhân suy tim do BCTPĐ, một số đặc điểm được thêm vào để định nghĩa lại đánh giá nguy cơ của đột quỵ do tim [7], [23], [36], nhưng giá trị tiên đoán so với HCM Risk-SCD vẫn chưa biết rõ. Đáng lưu ý, HCM Risk-SCD chưa được chuẩn hoá ở những bệnh nhân BCTPĐ do nguyên nhân chuyển hoá/dự trữ, hoặc sau cắt thành tim/chích đốt vách.
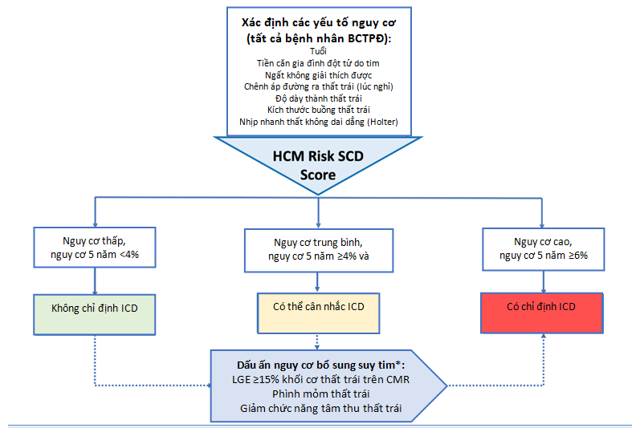
Hình 3. Đánh giá nguy cơ đột tử do tim ở những bệnh nhân suy tim và BCTPĐ
BCTPĐ: bệnh cơ tim phì đại; ICD: máy phá rung chuyển nhịp cấy được; LGE, late gadolinium enhancement, tăng cản từ muộn; *: Ở những bệnh nhân nguy cơ thấp đến trung bình, ICD có thể được khuyến cáo nếu có những dấu ấn của tăng nguy cơ đột tử do tim sau khi cân nhắc cẩn thận các biến chứng có thể xảy ra.
Những bệnh nhân BCTPĐ không tắc nghẽn có phân suất tống máu thất trái giảm (< 50%) nên được điều trị theo khuyến cáo của suy tim có phân suất tống máu giảm [15]. Trong bối cảnh rối loạn chức năng thất trái tiến triển, triệu chứng suy tim kháng trị và block nhánh trái, có ít dữ liệu ủng hộ đặt CRT ở những bệnh nhân phân suất tống máu thất trái < 50% [61], trong khi những bệnh nhân với phân suất tống máu thất trái ≤ 35% và block nhánh trái nên được cân nhắc đặt CRT theo các hướng dẫn điều trị suy tim hiện hành [57].
Ở những bệnh nhân BCTPĐ và suy tim nặng, hỗ trợ tuần hoàn cơ học dài hạn hiếm khi là cân nhắc thích hợp cho giai đoạn bắt cầu để ghép tim, do kích thước buồng thất trái nhỏ và rối loạn đổ đầy nặng. Tuy nhiên, một nghiên cứu nhỏ cho thấy cải thiện kết cục ở những bệnh nhân BCTPĐ với thiết bị hỗ trợ thất trái so với những bệnh nhân bệnh cơ tim dãn nở, nhưng có nguy cơ biến chứng cao hơn [73]. Ghép tim nên được cân nhắc ở những bệnh nhân tiến triển đến suy tim nặng dù đã điều trị theo khuyến cáo. Vào thời điểm ghép tim, hầu hết bệnh nhân đều có rối loạn chức năng tâm thu thất trái đáng kể. Một tỉ lệ nhỏ bệnh nhân BCTPĐ có thể cần phải ghép tim vì suy tim nặng dù phân suất tống máu thất trái bảo tồn[63].
Điều trị những bệnh nhân với các rối loạn dự trữ
Ở những bệnh nhân BCTPĐ do bệnh Anderson-Fabry, điều trị thay thế enzym hiệu quả với agalsidase-α và agalsidase-β [41], [2], [13], hoặc với uống chaperone, miagalastat [25], [45], nên được khởi đầu sớm khi có thể. Ở những bệnh nhân bệnh Pompe, (bệnh tích tụ glycogen type II), điều trị thay thế hormone với α-glucosidase người tái tổ hợp được sử dụng (Bảng 1) [58], [6]. Vì không có điều trị chuyên biệt cho những bệnh nhân bệnh Danon, theo dõi sát được khuyến cáo do bệnh cảnh ác tính của bệnh, bao gồm có cấy ICD ngưỡng thấp và lên danh sách sớm cho ghép tim ở những đối tượng thích hợp [10].
Bảng 1. Biện pháp điều trị của các rối loạn dự trữ
| Bệnh Anderson-Fabry | |
| Điều trị thay thế enzyme: agalsidase-α hoặc agalsidase-β | · Giảm chỉ số khối cơ thất trái và tăng có ý nghĩa phân suất rút ngắn vùng giữa thành tim
· Cải thiện kết cục gộp tim, thận và mạch máu não hoặc tử vong · Hiệu quả điều trị giảm một phần do hình thành kháng thể có thể cải thiện bởi các chất điều hoà miễn dịch hoặc phối hợp với chaperone uống |
| Uống chaperon: migalstat | · Ảnh hưởng tương tự lên kết cục thận, tim mạch và mạch máu não so với điều trị thay thế enzyme.
· Có thể ảnh hưởng tích cực lên xơ hoá và phì đại thất trái |
| Bệnh Pompe | |
| Điều trị thay thế enzyme: α-glucosidase | · Làm giảm phì đại thất trái (nếu được điều trị sớm trong diễn tiến của bệnh) |
KẾT LUẬN
Bệnh cơ tim phì đại là hậu quả của đột biến gen sarcomere, rối loạn dự trữ, hội chứng di truyền và rối loạn thần kinh cơ, ty thể. BCTPĐ chiếm khoảng 2-3% các trường hợp suy tim. Điều trị suy tim do BCTPĐ cần tuân thủ các biện pháp điều trị chung theo các hướng dẫn thực hành lâm sàng hiện hành về suy tim và điều trị nguyên nhân đặc hiệu để làm giảm triệu chứng, cải thiện chất lượng cuộc sống, tránh tái nhập viện và phòng ngừa nguy cơ đột tử và tử vong cho bệnh nhân.
TÀI LIỆU THAM KHẢO
- 1. Akhtar M. M., Elliott P. M. (2018), “Anderson-Fabry disease in heart failure”, Biophys Rev, 10 (4), 1107-1119.
- Banikazemi M., Bultas J., Waldek S., et al. (2007), “Agalsidase-beta therapy for advanced Fabry disease: a randomized trial”, Ann Intern Med, 146 (2), 77-86.
- Biagini E., Spirito P., Rocchi G., et al. (2009), “Prognostic implications of the Doppler restrictive filling pattern in hypertrophic cardiomyopathy”, Am J Cardiol, 104 (12), 1727-31.
- Briguori C., Betocchi S., Romano M., et al. (1999), “Exercise capacity in hypertrophic cardiomyopathy depends on left ventricular diastolic function”, Am J Cardiol, 84 (3), 309-15.
- Calcagnino M., O’Mahony C., Coats C., et al. (2011), “Exercise-induced left ventricular outflow tract obstruction in symptomatic patients with Anderson-Fabry disease”, J Am Coll Cardiol, 58 (1), 88-9.
- Capelle Carine, Poelman Esther, Frohn-Mulder Ingrid, et al. (2018), “Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase”, International Journal of Cardiology, 269.
- Chan R. H., Maron B. J., Olivotto I., et al. (2014), “Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy”, Circulation, 130 (6), 484-95.
- Charron P., Elliott P. M., Gimeno J. R., et al. (2018), “The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: baseline data and contemporary management of adult patients with cardiomyopathies”, Eur Heart J, 39 (20), 1784-1793.
- Corrado D., Basso C., Schiavon M., et al. (1998), “Screening for hypertrophic cardiomyopathy in young athletes”, N Engl J Med, 339 (6), 364-9.
- D’Souza R S., Levandowski C., Slavov D., et al. (2014), “Danon disease: clinical features, evaluation, and management”, Circ Heart Fail, 7 (5), 843-9.
- Desai M. Y., Bhonsale A., Smedira N. G., et al. (2013), “Predictors of long-term outcomes in symptomatic hypertrophic obstructive cardiomyopathy patients undergoing surgical relief of left ventricular outflow tract obstruction”, Circulation, 128 (3), 209-16.
- Efthimiadis G. K., Pagourelias E. D., Parcharidou D., et al. (2013), “Clinical characteristics and natural history of hypertrophic cardiomyopathy with midventricular obstruction”, Circ J, 77 (9), 2366-74.
- El Dib R., Gomaa H., Ortiz A., et al. (2017), “Enzyme replacement therapy for Anderson-Fabry disease: A complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies”, PLoS One, 12 (3), e0173358.
- Elliott P., Andersson B., Arbustini E., et al. (2008), “Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases”, Eur Heart J, 29 (2), 270-6.
- Elliott P. M., Anastasakis A., Borger M. A., et al. (2014), “2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC)”, Eur Heart J, 35 (39), 2733-79.
- Faber L., Welge D., Fassbender D., et al. (2007), “One-year follow-up of percutaneous septal ablation for symptomatic hypertrophic obstructive cardiomyopathy in 312 patients: predictors of hemodynamic and clinical response”, Clin Res Cardiol, 96 (12), 864-73.
- Ferrantini C., Belus A., Piroddi N., et al. (2009), “Mechanical and energetic consequences of HCM-causing mutations”, J Cardiovasc Transl Res, 2 (4), 441-51.
- Galati G., Leone O., Pasquale F., et al. (2016), “Histological and Histometric Characterization of Myocardial Fibrosis in End-Stage Hypertrophic Cardiomyopathy: A Clinical-Pathological Study of 30 Explanted Hearts”, Circ Heart Fail, 9 (9).
- Garcia-Pavia P., Vazquez M. E., Segovia J., et al. (2011), “Genetic basis of end-stage hypertrophic cardiomyopathy”, Eur J Heart Fail, 13 (11), 1193-201.
- Girolami F., Ho C. Y., Semsarian C., et al. (2010), “Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations”, J Am Coll Cardiol, 55 (14), 1444-53.
- Green J. J., Berger J. S., Kramer C. M., et al. (2012), “Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy”, JACC Cardiovasc Imaging, 5 (4), 370-7.
- Hada Y., Sakamoto T., Amano K., et al. (1987), “Prevalence of hypertrophic cardiomyopathy in a population of adult Japanese workers as detected by echocardiographic screening”, Am J Cardiol, 59 (1), 183-4.
- Harris K. M., Spirito P., Maron M. S., et al. (2006), “Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy”, Circulation, 114 (3), 216-25.
- Ho C. Y. (2009), “Hypertrophic cardiomyopathy: preclinical and early phenotype”, J Cardiovasc Transl Res, 2 (4), 462-70.
- Hughes D. A., Nicholls K., Shankar S. P., et al. (2017), “Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study”, J Med Genet, 54 (4), 288-296.
- Kuhn H., Lawrenz T., Lieder F., et al. (2008), “Survival after transcoronary ablation of septal hypertrophy in hypertrophic obstructive cardiomyopathy (TASH): a 10 year experience”, Clin Res Cardiol, 97 (4), 234-43.
- Kuperstein R., Klempfner R., Ofek E., et al. (2018), “De novo mitral regurgitation as a cause of heart failure exacerbation in patients with hypertrophic cardiomyopathy”, Int J Cardiol, 252, 122-127.
- Limongelli G., Masarone D., Frisso G., et al. (2017), “Clinical and genetic characterization of patients with hypertrophic cardiomyopathy and right atrial enlargement”, J Cardiovasc Med (Hagerstown), 18 (4), 249-254.
- Linhart A., Kampmann C., Zamorano J. L., et al. (2007), “Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey”, Eur Heart J, 28 (10), 1228-35.
- Losi M. A., Nistri S., Galderisi M., et al. (2010), “Echocardiography in patients with hypertrophic cardiomyopathy: usefulness of old and new techniques in the diagnosis and pathophysiological assessment”, Cardiovasc Ultrasound, 8, 7.
- Magdi H. Yacoub, Iacopo Olivotto, Cecchi Franco (2007), “‘End-Stage’ Hypertrophic Cardiomyopathy: From Mystery to Model”, Nat Clin Pract Cardiovasc Med, 4 (5), 232-233.
- Maron B. J., Maron M. S., Wigle E. D., et al. (2009), “The 50-year history, controversy, and clinical implications of left ventricular outflow tract obstruction in hypertrophic cardiomyopathy from idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy: from idiopathic hypertrophic subaortic stenosis to hypertrophic cardiomyopathy”, J Am Coll Cardiol, 54 (3), 191-200.
- Maron B. J., Roberts W. C., Arad M., et al. (2009), “Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy”, Jama, 301 (12), 1253-9.
- Maron B. J., Rowin E. J., Casey S. A., et al. (2015), “Hypertrophic Cardiomyopathy in Adulthood Associated With Low Cardiovascular Mortality With Contemporary Management Strategies”, J Am Coll Cardiol, 65 (18), 1915-28.
- Maron B. J., Rowin E. J., Udelson J. E., et al. (2018), “Clinical Spectrum and Management of Heart Failure in Hypertrophic Cardiomyopathy”, JACC Heart Fail, 6 (5), 353-363.
- Maron M. S., Finley J. J., Bos J. M., et al. (2008), “Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy”, Circulation, 118 (15), 1541-9.
- Maron M. S., Olivotto I., Betocchi S., et al. (2003), “Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy”, N Engl J Med, 348 (4), 295-303.
- Maron M. S., Olivotto I., Maron B. J., et al. (2009), “The case for myocardial ischemia in hypertrophic cardiomyopathy”, J Am Coll Cardiol, 54 (9), 866-75.
- Maron M. S., Olivotto I., Zenovich A. G., et al. (2006), “Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction”, Circulation, 114 (21), 2232-9.
- Maron M. S., Rowin E. J., Olivotto I., et al. (2016), “Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy”, J Am Coll Cardiol, 67 (12), 1399-1409.
- Mehta A., Beck M., Elliott P., et al. (2009), “Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data”, Lancet, 374 (9706), 1986-96.
- Mehta A., Ricci R., Widmer U., et al. (2004), “Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey”, Eur J Clin Invest, 34 (3), 236-42.
- Melacini P., Basso C., Angelini A., et al. (2010), “Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy”, Eur Heart J, 31 (17), 2111-23.
- Millat G., Bouvagnet P., Chevalier P., et al. (2010), “Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy”, Eur J Med Genet, 53 (5), 261-7.
- Muntze J., Salinger T., Gensler D., et al. (2018), “Treatment of hypertrophic cardiomyopathy caused by cardiospecific variants of Fabry disease with chaperone therapy”, Eur Heart J, 39 (20), 1861-1862.
- Nistri S., Olivotto I., Betocchi S., et al. (2006), “Prognostic significance of left atrial size in patients with hypertrophic cardiomyopathy (from the Italian Registry for Hypertrophic Cardiomyopathy)”, Am J Cardiol, 98 (7), 960-5.
- Nistri S., Thiene G., Basso C., et al. (2003), “Screening for hypertrophic cardiomyopathy in a young male military population”, Am J Cardiol, 91 (8), 1021-3, a8.
- O’Hanlon R., Grasso A., Roughton M., et al. (2010), “Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy”, J Am Coll Cardiol, 56 (11), 867-74.
- Olivotto I., Cecchi F., Casey S. A., et al. (2001), “Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy”, Circulation, 104 (21), 2517-24.
- Olivotto I., Cecchi F., Poggesi C., et al. (2012), “Patterns of disease progression in hypertrophic cardiomyopathy: an individualized approach to clinical staging”, Circ Heart Fail, 5 (4), 535-46.
- Olivotto I., Girolami F., Ackerman M. J., et al. (2008), “Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy”, Mayo Clin Proc, 83 (6), 630-8.
- Olivotto I., Girolami F., Nistri S., et al. (2009), “The many faces of hypertrophic cardiomyopathy: from developmental biology to clinical practice”, J Cardiovasc Transl Res, 2 (4), 349-67.
- Olivotto I., Maron B. J., Appelbaum E., et al. (2010), “Spectrum and clinical significance of systolic function and myocardial fibrosis assessed by cardiovascular magnetic resonance in hypertrophic cardiomyopathy”, Am J Cardiol, 106 (2), 261-7.
- Ommen S. R., Maron B. J., Olivotto I., et al. (2005), “Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy”, J Am Coll Cardiol, 46 (3), 470-6.
- Pasqualucci D., Fornaro A., Castelli G., et al. (2015), “Clinical Spectrum, Therapeutic Options, and Outcome of Advanced Heart Failure in Hypertrophic Cardiomyopathy”, Circ Heart Fail, 8 (6), 1014-21.
- Patel V., O’Mahony C., Hughes D., et al. (2015), “Clinical and genetic predictors of major cardiac events in patients with Anderson-Fabry Disease”, Heart, 101 (12), 961-6.
- Ponikowski P., Voors A. A., Anker S. D., et al. (2016), “2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC”, Eur Heart J, 37 (27), 2129-2200.
- Prater S. N., Banugaria S. G., DeArmey S. M., et al. (2012), “The emerging phenotype of long-term survivors with infantile Pompe disease”, Genet Med, 14 (9), 800-10.
- Qintar M., Morad A., Alhawasli H., et al. (2012), “Pacing for drug-refractory or drug-intolerant hypertrophic cardiomyopathy”, Cochrane Database Syst Rev, (5), Cd008523.
- Rastegar H., Boll G., Rowin E. J., et al. (2017), “Results of surgical septal myectomy for obstructive hypertrophic cardiomyopathy: the Tufts experience”, Ann Cardiothorac Surg, 6 (4), 353-363.
- Rogers D. P., Marazia S., Chow A. W., et al. (2008), “Effect of biventricular pacing on symptoms and cardiac remodelling in patients with end-stage hypertrophic cardiomyopathy”, Eur J Heart Fail, 10 (5), 507-13.
- Rosmini S., Biagini E., O’Mahony C., et al. (2017), “Relationship between aetiology and left ventricular systolic dysfunction in hypertrophic cardiomyopathy”, Heart, 103 (4), 300-306.
- Rowin E. J., Maron B. J., Kiernan M. S., et al. (2014), “Advanced heart failure with preserved systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant”, Circ Heart Fail, 7 (6), 967-75.
- Schäfers Michael, Dutka David, Rhodes Christopher G., et al. (1998), “Myocardial Presynaptic and Postsynaptic Autonomic Dysfunction in Hypertrophic Cardiomyopathy”, Circulation Research, 82 (1), 57-62.
- Seydelmann N., Liu D., Kramer J., et al. (2016), “High-Sensitivity Troponin: A Clinical Blood Biomarker for Staging Cardiomyopathy in Fabry Disease”, J Am Heart Assoc, 5 (6).
- Shah J. S., Lee P., Hughes D., et al. (2005), “The natural history of left ventricular systolic function in Anderson-Fabry disease”, Heart, 91 (4), 533-4.
- Sherrid M. V., Balaram S., Kim B., et al. (2016), “The Mitral Valve in Obstructive Hypertrophic Cardiomyopathy: A Test in Context”, J Am Coll Cardiol, 67 (15), 1846-1858.
- Sherrid M. V., Barac I., McKenna W. J., et al. (2005), “Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy”, J Am Coll Cardiol, 45 (8), 1251-8.
- Sorajja P., Ommen S. R., Nishimura R. A., et al. (2003), “Myocardial bridging in adult patients with hypertrophic cardiomyopathy”, J Am Coll Cardiol, 42 (5), 889-94.
- Tardiff J. C. (2005), “Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes”, Heart Fail Rev, 10 (3), 237-48.
- Thaman R., Gimeno J. R., Murphy R. T., et al. (2005), “Prevalence and clinical significance of systolic impairment in hypertrophic cardiomyopathy”, Heart, 91 (7), 920-5.
- Thorvaldsen T., Benson L., Dahlstrom U., et al. (2016), “Use of evidence-based therapy and survival in heart failure in Sweden 2003-2012”, Eur J Heart Fail, 18 (5), 503-11.
- Topilsky Y., Pereira N. L., Shah D. K., et al. (2011), “Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy”, Circ Heart Fail, 4 (3), 266-75.
- Torralba-Cabeza M. A., Olivera S., Hughes D. A., et al. (2011), “Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease”, Mol Genet Metab, 104 (3), 301-7.
- Weidemann F., Niemann M., Herrmann S., et al. (2007), “A new echocardiographic approach for the detection of non-ischaemic fibrosis in hypertrophic myocardium”, Eur Heart J, 28 (24), 3020-6.
- Wu J. C., Ho C. Y., Skali H., et al. (2010), “Cardiovascular manifestations of Fabry disease: relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase A activity”, Eur Heart J, 31 (9), 1088-97.
- Yusen R. D., Edwards L. B., Dipchand A. I., et al. (2016), “The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Lung and Heart-Lung Transplant Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant”, J Heart Lung Transplant, 35 (10), 1170-1184.
- Zou Y., Song L., Wang Z., et al. (2004), “Prevalence of idiopathic hypertrophic cardiomyopathy in China: a population-based echocardiographic analysis of 8080 adults”, Am J Med, 116 (1), 14-8.


{kind=link}