


NHỮNG TIẾN BỘ PHÂN TẦNG NGUY CƠ ĐỘT TỬ TIM TRONG BỆNH CƠ TIM PHÌ ĐẠI
TS. PHẠM HỮU VĂN
Tóm tắt
SCD là biến chứng nguy hiểm nhất của bệnh cơ tim phì đại (HCM) và có tỷ lệ hàng năm là 1%. Việc ước tính nguy cơ chính xác của SCD có thể vô cùng khó khăn và gần đây nó đã trở thành nguồn tranh luận giữa các chuyên gia châu Âu và Mỹ. Mặc dù yếu tố nguy cơ chính của SCD là ngừng tim được cứu sống trước đây được xác định, nhưng việc xác định những cá nhân có nguy cơ cao bị SCD trong dự phòng nguyên phát vẫn còn là một vấn đề được tranh luận. Điều này là do thực tế các khuyến cáo dựa trên các nghiên cứu thuần tập quan sát, hồi cứu. Trước đây, 5 yếu tố nguy cơ được coi là có nguy cơ đối với SCD và sự hiện diện của 2 hoặc nhiều hơn trong số này được coi là chỉ định cho việc cấy ICD để phòng ngừa tiên phát, gồm tiền sử gia đình có SCD, ngất, độ dày thành thất trái tối đa (LV), nhịp nhanh thất tạm thời (NSVT), phản ứng huyết áp bất thường khi gắng sức. Các hướng dẫn của ACCF/AHA năm 2011 đã xác nhận lần này khuyến cáo bị giảm đi khi gán vai trò chính cho ba yếu tố nguy cơ đầu tiên. Ở bên kia biển Đại Tây Dương, hướng dẫn ESC gần đây nhất (2014) đã xác nhận một mô hình dự báo nguy cơ mới bao gồm: tuổi, tiền sử gia đình bị SCD, ngất không rõ nguyên nhân, tắc nghẽn đường ra LV, độ dày thành LV tối đa, đường kính tâm nhĩ trái, NSVT. Mô hình EU đã được xác nhận sau một nghiên cứu thuần tập hồi cứu, đa trung tâm trên 3675 bệnh nhân và nó chính xác hơn đáng kể so với mô hình của Hoa Kỳ dựa trên các nhóm bệnh nhân cũ và nhỏ. Tuy nhiên, trong vài năm qua, nghiên cứu tim mạch đang tiến rất nhanh trong việc phát hiện ra các yếu tố nguy cơ sửa đổi mới. Do đó, phình mỏm thất trái (LV), đa đột biến gen sarcomere, xơ hóa cơ tim (Myocardial fibrosis: MF) và giai đoạn cuối (ES) HCM ngày càng trở nên quan trọng, đặc biệt ở những trường hợp có nguy cơ trung bình. Do đó, một số lượng lớn các nghiên cứu và hai phân tích tổng hợp đã được công bố trên MF, cho thấy một yếu tố dự báo nguy cơ mạnh độc lập về tỷ lệ tử vong do tim mạch và của SCD. Đặc biệt, một nghiên cứu gần đây cho thấy mức độ LGE ≥15% (Late Gadolinium Enhancement: LGE [gia tăng muộn Gadolinum]) khối lượng LV làm tăng > 2 lần nguy cơ SCD và lượng LGE ≥20% làm tăng > 3 lần nguy cơ phát triển ESHCM.
Cuối cùng, một nghiên cứu cơ bản đa trung tâm ở châu Âu đã cung cấp đánh giá mô học định lượng chi tiết đầu tiên về mức độ, sự phân bố, các kiểu và các loại MF trong một quần thể lớn các bệnh tim mạch ES-HCM. Hơn nữa, nó đánh giá mối quan hệ định lượng giữa đánh giá LGE-CMR và phân tích mô học của MF. Nghiên cứu đã khám phá những quan điểm mới về MF, về đánh giá hình ảnh của nó và cuối cùng là về sinh lý bệnh của HCM và của SCD.
Từ khóa: bệnh cơ tim phì đại, đột tử do tim, các yếu tố nguy cơ, xơ hóa cơ tim
MỞ ĐẦU
HCM được xác định bằng sự hiện diện của phì đại cơ tim (nghĩa là, độ dày thành ≥15 mm ở một hoặc nhiều đoạn cơ tim thất trái (LV) được đo bằng bất kỳ kỹ thuật hình ảnh nào – siêu âm tim, chụp cộng hưởng từ tim (CMR) hoặc chụp cắt lớp vi tính) khi không có đủ sự căng thẳng huyết động để giải thích mức độ phì đại (như bệnh van tim, tăng huyết áp, bệnh tim bẩm sinh) và các bệnh toàn thân như bệnh amyloidosis và bệnh dự trữ glycogen có thể là nguyên nhân làm tăng độ dày cơ tim (Elliott, 2014., Elliott, 2008.).
Ở trẻ em, chẩn đoán HCM yêu cầu độ dày thành LV lớn hơn hai độ lệch chuẩn so với giá trị trung bình dự đoán (điểm z > 2, trong đó điểm số z được định nghĩa là số độ lệch chuẩn so với trung bình dân số) (Elliott, Năm 2014.).
HCM là bệnh tim di truyền thường xuyên nhất, với tỷ lệ hiện mắc ước tính là 1/500 người tương ứng với 0,2% dân số toàn cầu. Tuy nhiên, các nghiên cứu quần thể di truyền gần đây nhất báo cáo tỷ lệ hiện hành cao hơn 1 trên 200 người (Semsarian, 2015).
Ở hơn 60% thanh thiếu niên và người lớn bị HCM, bệnh lây lan do một hoặc nhiều đột biến của một hoặc nhiều gen protein sarcomere ở tim (Elliott, 2014.). Hơn nữa, điều quan trọng cần lưu ý là nhiều bệnh di truyền và không di truyền tim khác có thể giống kiểu hình sarcomeric HCM (5-10% trường hợp). Đây được gọi là “kiểu hình copy (phenocopies) của sarcomeric HCM” (Hình 1).
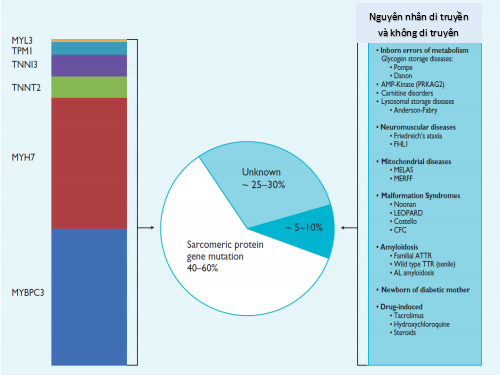
Hình 1. Các bệnh căn khác nhau của HCM (Elliott 2014)
Inborn errors of metabolism: các sai sót chuyển hóa bẩm sinh. Glycogen storage disease: bệnh lưu trữ glycogen. Carnitime disorders: các rối loạn carnitine. Lysosomal storage disease: bệnh lưu trữ lysosome. Neuromuscular diseases: Các bệnh thần kinh cơ. Friedreich’s ataxia: thất điểu Friedreich. Mitochondrial diseases: các bệnh của mitochondri. Amyloidosis: bệnh amyloidose. Newborn of diabetic mother: trẻ sơ sinh của người mẹ tiểu đường. Drug – induced: được tạo ra do thuốc. Unknown: không rõ. Sarcomeric protein gene mutation: Đột biến gen sarcoma protein.
Bệnh sử tự nhiên của HCM là rất nhiều nguồn gốc. Đại đa số bệnh nhân không có triệu chứng và có tuổi thọ bình thường (gần 70%). Nhiều người khác phát triển các biến chứng chính của HCM đó là: rung nhĩ và các biến cố huyết khối tắc mạch, viêm nội tâm mạc nhiễm trùng (ở bệnh nhân HCM tắc nghẽn), SCD, HCM giai đoạn cuối (end-stage HCM: ESHCM) và suy tim tiến triển.
Mặc dù một số tư liệu gần đây báo cáo tỷ lệ tử vong hàng năm <1% khi sử dụng các thiết bị điều trị hiện đại, (máy khử rung tim: ICD), nghép tim và dự phòng đột quỵ), tỷ lệ các biến cố đe dọa tính mạng hoặc tàn phế mỗi năm vẫn ở mức cao. Thật vậy, trong những nhóm thuần tập đó, gần 15% bệnh nhân đột tử hoặc được sốc ICD thích hợp hoặc suy tim tiến triển. Ngoài ra, 19% bệnh nhân phát triển rung nhĩ hoặc đột quỵ (Elliott, 2016., Maron, 2016.).
Do đó, cả SCD và HCM giai đoạn cuối đều là những biến chứng đáng ngại nhất, và chúng ta có thể tóm tắt khái niệm này trong Hình 2.
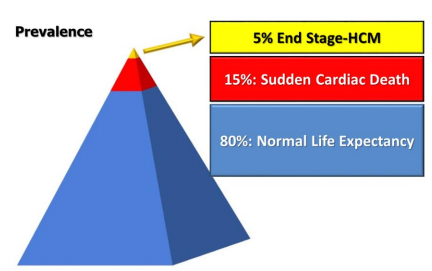
Hình 2. Tỷ lệ mắc hai biến chứng đáng ngại nhất trong HCM.
Prevalence: tỷ lệ. End Stage-HCM: giai đoạn cuối của bệnh cơ tim phì đại. Sudden Cardiac Death: Đột tử tim. Normal Life Expectancy: Tuổi thọ bình thường.
* Phân tầng nguy cơ SCD trong bệnh cơ tim phì đại trạng thái kỹ thuật hiện nay
SCD được định nghĩa là tử vong tự nhiên do nguyên nhân tim, xảy ra bất ngờ trong vòng 1 giờ kể từ khi xuất hiện các triệu chứng mới hoặc là tử vong ngoài sự mong muốn và không lường trước được, ngoại trừ khi một nguyên nhân tử vong không do tim cụ thể đã được tìm thấy. Các yếu tố chính là: tự nhiên, nhanh chóng và bất ngờ.
SCD là biến chứng khủng khiếp và tàn khốc nhất của HCM và các nghiên cứu hiện đại nhất báo cáo tỷ lệ mắc hàng năm ước tính là 1% (Elliott, 2014., Gersh, 2011., Weissler-Snir, 2016.).
Nguyên nhân chính của SCD là các biến cố loạn nhịp gây tử vong như rung thất, nhịp nhanh thất đa hình và hoạt động điện vô mạch.
Bảng 1. Đánh giá nguy cơ SCD ở các bệnh nhân HCM
| Thước đo | Manh mối được kết hợp với SCD |
| Bệnh sử lâm sàng | • Có bệnh sử cá nhân ngừng tim được cứu sống (không có bệnh sử bệnh mạch vành, bệnh van tim v.v.…)
• Tiền sử SCD gia đình (một hoặc nhiều người thân thế hệ đầu) • Ngất không giải thích được. |
| ECG 24h | • Nhịp nhanh thất tạm thời (NSVT) |
| Siêu âm qua thành ngực (khi nghỉ hoặc gắng sức) | • Độ dày thành LV tối đa ≥ 30 mm
• Tắc nghẽn LVOT • Đường kính tâm nhĩ trái • Phình mỏm LV • LVEF ≤50% (tức là tiến triển Giai đoạn cuối) |
| Cộng hưởng Từ Tim | • Độ dày thành LV tối đa ≥ 30 mm
• Đường kính tâm nhĩ trái • Xơ hóa cơ tim ≥15% • Phình mỏm LV • LVEF≤50% (tức là tiến triển Giai đoạn cuối) |
| Test gắng sức | • Đáp ứng huyết áp với gắng sức bất thường |
| Phân tích DNA (NGS DNA) | • Đột biến riêng biệt?
• Đột biến kép hay đột biến ba? |
Ước tính nguy cơ SCD là một trong những khía cạnh quan trọng nhất của việc quản lý bệnh nhân bị HCM ảnh hưởng và đòi hỏi một đánh giá toàn diện, tỉ mỉ và phù hợp (Bảng 1) Do đó, ước tính nguy cơ SCD chính xác có thể vô cùng khó khăn và gần đây đã trở thành nguồn tranh luận giữa các chuyên gia châu Âu và Mỹ. Mặc dù yếu tố nguy cơ chính của SCD chắc chắn là đã một lần ngừng tim đã được hồi sức trước đó, việc xác định các cá nhân có nguy cơ cao bị SCD trong dự phòng tiên phát vẫn còn là một vấn đề còn tranh cãi.
Có điều này là do thực tế không có thử nghiệm ngẫu nhiên hoặc mô hình dự báo tiền cứu được xác nhận về mặt thống kê trong lĩnh vực này để các khuyến nghị dựa trên các nghiên cứu thuần tập quan sát, hồi cứu.
Trong lịch sử, 5 yếu tố nguy cơ được coi là có nguy cơ đối với SCD và sự hiện diện của 2 hoặc nhiều hơn trong số này được coi là dấu hiệu cho việc cấy ICD để phòng ngừa tiên phát (Elliott, 2014., Gersh, 2011., Weissler-Snir, 2016.), I E,
- Tiền sử gia đình có SCD
- Ngất không giải thích được
- Độ dày thành tâm thất trái (LV) tối đa ≥ 30 mm
- Nhịp nhanh thất tạm thời (NSVT)
- Phản ứng huyết áp bất thường khi gắng sức
* Thái độ của Y học
Các hướng dẫn của ACCF/AHA năm 2011 đã xác nhận khuyến cáo này không còn phù hợp, đã hết thời gian và dựa trên bằng chứng không đầy đủ về việc cấy ICD để phòng ngừa tiên phát.
Tuy nhiên, đối với hướng dẫn chung ACC/ESC 2003 trước đây, trong đó sự hiện diện của hai hoặc nhiều yếu tố nguy cơ ở bệnh nhân HCM được coi là một chỉ định cấy ICD để phòng ngừa tiên phát, các chuyên gia ACCF/ HA năm 2011 đã được chỉ định vai trò chính của ba yếu tố nguy cơ đầu tiên cũng có thể xuất hiện đơn lẻ (Gersh, 2011.). Thật vậy, một số liệu đăng ký quốc tế gồm 506 bệnh nhân cho thấy trong số bệnh nhân HCM được cấy ICD, số lượng các yếu tố nguy cơ không tương quan với tỷ lệ các lần sốc phù hợp tiếp theo của thiết bị (Maron, 2008). Tuy nhiên, điều quan trọng cần lưu ý là đây là bằng chứng rất yếu và có tính hồi cứu về một nhóm nhỏ bệnh nhân và các lần sốc ICD không phải là đại diện chính xác của SCD; do đó, việc tổng hợp kết quả từ các nhóm ICD được chọn lọc cao này cho dân số HCM nói chung là điều đáng nghi ngờ và trái với kết quả của các nhóm lớn hơn, không được chọn lọc trong thuần tập HCM.
Việc nâng cấp ba yếu tố đầu tiên như những yếu tố chính đủ để cấy ICD không được hỗ trợ của bất kỳ bằng chứng quan trọng mới hoặc nghiên cứu tiền cứu nào.
Hơn nữa, một nghiên cứu xác nhận gần đây đã có thể chỉ ra tập hợp các yếu tố nguy cơ đơn lẻ có tương quan với SCD nhiều hơn một yếu tố nguy cơ và mức độ liên quan của hai hoặc nhiều yếu tố nguy cơ với SCD lớn hơn so với một yếu tố nguy cơ duy nhất (O ‘ Mahony, 2011.). Mặt khác, nghiên cứu này cho thấy tỷ lệ mắc SCD ở những bệnh nhân có một yếu tố nguy cơ đơn lẻ không khác biệt đáng kể so với những bệnh nhân không có yếu tố nguy cơ (O’Mahony, 2011.).
Cách tiếp cận lịch sử có một số hạn chế đáng kể vì nó dựa trên các nghiên cứu hồi cứu cũ với nhóm nhỏ bệnh nhân chỉ ước tính nguy cơ tương đối (chứ không phải tuyệt đối) và một số yếu tố nguy cơ – chẳng hạn như độ dày thành LV – được coi là biến nhị phân khi chúng có liên quan đến sự gia tăng liên tục nguy cơ.
Như vậy, hướng dẫn của ACCF/AHA 2011 có thể được xác định một cách dễ dàng và chính xác là các ý kiến và khuyến cáo của chuyên gia Hoa Kỳ về đánh giá nguy cơ SCD, nhưng không thể khẳng định các khuyến cáo đó là dựa trên bằng chứng.
Các tư liệu cho thấy việc tuân thủ nghiêm ngặt mô hình Bắc Mỹ có thể dẫn đến cấy ICD cho tới 40-60% bệnh nhân HCM với tỷ lệ sốc ICD thích hợp hàng năm thấp khoảng 2% trong nhóm thuần tập lớn trong HCM có cấy ICD (O’Mahony, 2011., Maron, 2015., O’Mahony, 2012.).
Cuối cùng, điều quan trọng cần lưu ý là quan điểm của Hoa Kỳ hiện đang dễ tiếp nhận các yếu tố nguy cơ sửa đổi mới hơn quan điểm của châu Âu.
* Quan điểm của châu Âu
Ở châu Âu, các hướng dẫn gần đây nhất (2014) của ESC đã xác nhận một mô hình dự đoán nguy cơ mới gồm 7 biến số (Elliott, 2014., O’Mahony, 2014.), đó là:
- Tuổi
- Tiền sử gia đình về SCD
- Ngất không rõ nguyên nhân
- Tắc nghẽn đường ra LV
- Độ dày thành LV tối đa
- Đường kính tâm nhĩ trái
- Nhịp nhanh thất tạm thời (NSVT)
Lần đầu tiên, các chuyên gia Châu Âu đã thực hiện một thay đổi đáng kể trong phân tầng nguy cơ của SCD ở HCM, để một số tác giả định nghĩa đây là “một cuộc cách mạng” và một số tác giả khác là “một sự tiến hóa quan trọng”.
Mô hình EU đã được xác nhận sau một nghiên cứu thuần tập hồi cứu đa trung tâm trên 3675 bệnh nhân – được xác định là HCM Nguy cơ Đột tử tim. Mô hình này cho điểm tiên lượng và có thể truy cập được như một máy tính trực tuyến. HCM nguy cơ đột tử tim sử dụng các biến có liên quan đến việc tăng nguy cơ SCD trong ít nhất một phân tích đa biến được công bố. Chỉ có bốn trong số năm yếu tố nguy cơ cổ điển vượt qua kiểm tra thống kê của ESC và được chứng minh là dựa trên bằng chứng khoa học. Hơn nữa, mô hình cung cấp ước tính nguy cơ SCD 5 năm riêng biệt và so sánh trực tiếp với mô hình sử dụng bốn yếu tố nguy cơ chính – rất giống với mô hình bệnh sử – hiệu suất của mô hình dự đoán được cải thiện đáng kể (chỉ số C từ 0,54 đến 0,7). HCM nguy cơ đột tử tim so sánh thuận lợi với các thuật toán dự đoán khác như CHA2DS2-VASC nổi tiếng.
Hướng dẫn của ESC HCM 2014 xác định ba vùng nguy cơ, đó là:
- Nguy cơ thấp: nguy cơ 5 năm <4%
- Nguy cơ trung gian: nguy cơ 5 năm ≥ 4% <6%
- Nguy cơ cao: nguy cơ 5 năm ≥ 6%
Những bệnh nhân có nguy cơ cao có khuyến cáo class IIa (“nên được xem xét”) để cấy ICD, trong khi những bệnh nhân có nguy cơ trung bình có khuyến nghị class IIb (“có thể được xem xét”) để cấy ICD. ICD thường không được chỉ định ở những bệnh nhân có nguy cơ thấp.
Mô hình châu Âu có khả năng giảm cấy ICD không cần thiết và có khả năng gây hại ở những bệnh nhân không bị SCD và xác định chính xác phần lớn những người có nhiều khả năng được hưởng lợi từ ICD.
Tại thời điểm này, điều quan trọng cần lưu ý là sự thay đổi hướng dẫn của ESC đã được thúc đẩy tốt và dựa trên bằng chứng rõ ràng và đưa ra một mô hình dự đoán chính xác hơn đáng kể so với mô hình của Hoa Kỳ dựa trên dân số cũ và ít bệnh nhân.
Cuối cùng, các hướng dẫn của ESC 2014 về HCM đã được cải thiện đáng kể phân tầng nguy cơ tiên lượng SCD và thể hiện một bước tiến trong hiểu biết hiện tại về nguy cơ SCD
* Các yếu tố nguy cơ SCD
- Tiền sử cá nhân về ngừng tim được cứu sống trước đây
Bệnh nhân HCM đã trải qua SCD hoặc VT kéo dài có nguy cơ cao nhất đối với các biến cố loạn nhịp tim ác tính tiếp theo. Tỷ lệ hàng năm gây ra các biến cố loạn nhịp ở nhóm bệnh nhân này là 10% (Cecchi, 1989., Elliott, 1999., Maron, 2009.). Như được thể hiện qua ba thử nghiệm [Thuốc chống loạn nhịp so với Máy khử rung tim (Antiarrhythmic drugs Versus Implantable Defibrillator: (AVID). Các nhà điều tra 1997), Nghiên cứu máy khử rung tim Canada (Canadian Implantable Defibrillator Study: CIDS) (Connolly, 2000.) và Nghiên cứu Hamburg về ngừng tim (Cardiac Arrest Study Hamburg: CASH) (Kuck, 2000.). được tiến hành ở những bệnh nhân đã bị ngừng tim hoặc loạn nhịp thất đe dọa tính mạng (nhịp nhanh thất không ổn định về huyết động kèm theo ngất) – không phân biệt bệnh tim nền – trong đó điều trị bằng ICD được so sánh với điều trị bằng thuốc chống loạn nhịp (chủ yếu là amiodaron), ICD giảm tỷ lệ tử vong do loạn nhịp tim. Hơn nữa, một phân tích tổng hợp của ba thử nghiệm đã chứng minh liệu pháp ICD có liên quan đến việc giảm 50% (95% CI 0,37, 0,67; P = 0,0001) tỷ lệ tử vong do loạn nhịp và giảm 28% (95% CI 0,60, 0,87; P = 0,006) giảm tổng số tử vong (Connolly, EHJ 2000.)
Do đó, những bệnh nhân đã từng bị ngừng tim hoặc loạn nhịp thất đe dọa tính mạng (VT dai dẳng) có chỉ định cấy ICD (Class I) cao nhất ở cả mô hình Châu Âu và Hoa Kỳ.
- Tuổi
Ảnh hưởng của tuổi tác lên SCD đã được khám phá trong nhiều nghiên cứu trong ba thập kỷ qua. Tuy nhiên, chỉ gần đây mối liên hệ đáng kể giữa ager càng trẻ và nguy cở SCD bị tăng lên đã được chứng minh bằng tư liệu trong 3 nghiên cứu (O’Mahony, 2014., Elliott, 1999., Spirito, 2009.). [Ager là gen trên nhiễm sắc thể 6p 21.3 mã hóa một thành viên của siêu họ globulin miễn dịch của các thụ thể bề mặt tế bào, không chỉ tương tác với các sản phẩm cuối cùng của quá trình glycosyl hóa nâng cao mà còn với các phân tử liên quan đến cân bằng nội môi, phát triển và viêm, cũng như bệnh Alzheimer và các bệnh khác]. Ngất không rõ nguyên nhân, NSVT và LVH nặng dường như quan trọng hơn khi liên quan đến tuổi trẻ. Mặt khác, những bệnh nhân bước vào thập kỷ thứ bảy của cuộc đời có nguy cơ SCD thấp ngay cả khi có liên quan đến một yếu tố nguy cơ kinh điển (Spirito, 2009.).
- Tiền sử gia đình về SCD
Một trong những quan sát đầu tiên trong bệnh cơ tim phì đại và nói chung trong các bệnh di truyền tim mạch là các biến cố SCD thường tập hợp trong gia đình. Thật không may, có một số dữ liệu mâu thuẫn về mối liên hệ này ở HCM do các định nghĩa khác nhau về SCD gia đình trong các nghiên cứu khác nhau. Tuy nhiên, tiền sử gia đình mắc SCD là rất quan trọng khi một hoặc nhiều người thân thế hệ 1 đột tử ở độ tuổi ≤ 40 tuổi có hoặc không có chẩn đoán HCM, hoặc khi người thân thế hệ 1 có chẩn đoán HCM đột ngột qua đời ở mọi lứa tuổi (Elliott, 2014). Một phân tích tổng hợp gần đây – bất chấp sự không đồng nhất về dân số của các nghiên cứu khác nhau – đã có thể chứng minh mối tương quan độc lập giữa tiền sử gia đình mắc SCD và nguy cơ SCD (Maron, 2013).
- Ngất không rõ nguyên nhân
Ngất (mất ý thức tạm thời thứ phát thoáng qua, giảm tưới máu não toàn thể) thường gặp ở bệnh nhân HCM – xảy ra ở ~ 20-25% bệnh nhân – nhưng là một chẩn đoán phân biệt lâm sàng đầy thách thức. HCM có nhiều nguyên nhân gây ngất: rung nhĩ, nhịp nhanh trên thất, loạn nhịp, loạn nhịp nhanh thất dai dẳng, tắc nghẽn LVOT liên quan đến gắng sức, phản ứng mạch máu bất thường, thiếu máu cục bộ cơ tim, ngất qua trung gian thần kinh (cường phế vị, ngất tình huống và ngất xong cảnh) và hạ huyết áp tư thế đứng. Sau khi chẩn đoán ngất, việc xác định nguyên nhân hoàn toàn dựa vào dữ liệu lịch sử do nhân chứng và bệnh nhân cung cấp.
Spirito và cộng sự. năm 2009 đã định nghĩa là ngất không rõ nguyên nhân “không rõ nguyên nhân, khi nó xảy ra trong những trường hợp không rõ ràng phù hợp với biến cố qua trung gian thần kinh, tức là, không có lời giải thích rõ ràng khi nghỉ ngơi hoặc trong các hoạt động bình thường hàng ngày, hoặc trong một nỗ lực cao độ.
Một số nghiên cứu sử dụng phân tích tỷ lệ sống sót đã chỉ ra mối liên hệ đáng kể giữa ngất không do thần kinh – không có lời giải thích sau khi điều tra – với nguy cơ SCD tăng lên (Christiaans, 2010., Elliott, 2000., Efthimiadis, 2009., Elliott, 2006., Gimeno, 2009, Dimitrow, 2010.).
Mối liên hệ mạnh nhất giữa ngất không rõ nguyên nhân và SCD xuất phát từ một nghiên cứu quốc tế đa trung tâm lớn trên 1511 bệnh nhân HCM (Spirito, 2009.). Trong nhóm thuần tập này, nguy cơ tương đối của SCD là 1,78 (khoảng tin cậy 95% 0,88 đến 3,51, P = 0,08) ở bệnh nhân ngất không rõ nguyên nhân và 0,91 (khoảng tin cậy 95% 0,00 đến 3,83, P = 1,0) ở những bệnh nhân ngất qua trung gian thần kinh so với với bệnh nhân không ngất. Đặc biệt, mối liên quan này chỉ có ý nghĩa ở những bệnh nhân ngất không rõ nguyên nhân trong vòng 6 tháng trước khi đánh giá ban đầu, cho thấy nguy cơ tăng gấp 5 lần đối với những bệnh nhân không ngất (tỷ số nguy cơ điều chỉnh 4,89, khoảng tin cậy 95% 2,19 đến 10,94). Bệnh nhân lớn tuổi (> 40 tuổi) có các đợt ngất từ xa (> 5 năm trước khi đánh giá ban đầu) không cho thấy tăng nguy cơ SCD (tỷ lệ nguy cơ đã điều chỉnh 0,38, khoảng tin cậy 95% 0,05 đến 2,74) (Spirito, 2009.).
- Tắc nghẽn đường ra LV
Một số nghiên cứu đã chứng minh tắc nghẽn đường ra thất trái (LV Outflow Tract: LVOT) là một yếu tố dự báo độc lập về kết quả bất lợi ở bệnh nhân HCM (Veselka, 2016., Maron, 2003., Sorajja, 2009., Elliott, 2006.). Những nghiên cứu này đã chứng minh tỷ lệ SCD cao hơn ở những bệnh nhân có độ chênh áp lúc nghỉ ≥ 30 mmHg. Điều quan trọng cần lưu ý là một nghiên cứu trước đây không chỉ cho thấy mối liên quan này mà còn cho thấy mối quan hệ giữa mức độ nghiêm trọng của tắc nghẽn và nguy cơ SCD (Elliott, 2006.). Thật vậy, tắc nghẽn đường ra thất trái không phải là một biến nhị phân mà nó có liên quan đến sự gia tăng liên tục nguy cơ (Elliott, 2006.). Do đó, lực cản càng cao thì nguy cơ SCD càng cao. Mô hình hoàn toàn bỏ qua mối quan hệ giữa tắc nghẽn LVOT và SCD và không tính đến nó để phân tầng nguy cơ SCD là không chính xác và gần đúng.
Sau khi xem xét lại các tư liệu, ESC quyết định đưa yếu tố nguy cơ này vào mô hình của mình và thử nghiệm nó trong nghiên cứu quốc tế đa trung tâm đã đề cập trước đó, xác nhận mối quan hệ đáng kể giữa tắc nghẽn LVOT (khi nghỉ ngơi và với kích thích Valsalva bất kể điều trị y tế đồng thời) và SCD và giữa mức độ tắc nghẽn LVOT và nguy cơ SCD (O’Mahony, 2014.).
Tuy nhiên, những nghi ngờ chính (hoặc vùng xám) về tắc nghẽn LVOT có tầm quan trọng tiên lượng của tắc nghẽn LVOT có thể có và tác động của điều trị tắc nghẽn LVOT (triệt phá vách bằng alcohol đối lại với điều trị nôi đối lại với lấy bỏ bằng ngoại khoa) đối với nguy cơ SCD.
- Độ dày thành LV tối đa
Mối liên quan giữa độ dày thành LV – độ dày lớn nhất ở vách trước, vách sau, thành bên và thành sau của LV, được đo ở mức van hai lá, cơ nhú và đỉnh bằng mặt phẳng trục ngắn cạnh sườn bằng siêu âm tim 2D – và nguy cơ SCD đã được ghi nhận rõ ràng trong một số nghiên cứu lớn, với độ dày thành LV ≥30 mm được coi là một yếu tố nguy cơ của SCD (Spirito, 2000., Elliott, 2001.).
Cũng như: Tuổi, tắc nghẽn LVOT và đường kính nhĩ trái, độ dày thành LV là một biến số liên tục và không phải là biến số nhị phân, do đó phì đại càng cao thì nguy cơ SCD càng cao (Elliott, 2014., O’Mahony, 2014.)
Điều quan trọng phải nhận ra nguy cơ không tự động tăng khi độ dày của tường đạt đến ngưỡng 30 mm, nhưng giống như trong tất cả các hiện tượng sinh học đều tăng theo kiểu tuyến tính, do đó độ dày LV 29 mm có liên quan đến rủi ro gần bằng liên quan đến 30 mm.
Tuy nhiên, có rất ít dữ liệu ở bệnh nhân phì đại cực đại (độ dày thành LV ≥35mm)
- Đường kính nhĩ trái (LA)
Hai nghiên cứu hồi cứu quốc tế đa trung tâm khác nhau đã ghi nhận mối liên hệ độc lập rõ ràng giữa đường kính LA và SCD (O’Mahony, 2014., Spirito, 2009.). Một trong số đó là nghiên cứu thuần tập hồi cứu được đề cập trước đó trên 3675 bệnh nhân HCM được thực hiện ở Châu Âu. Không có dữ liệu về mối liên hệ giữa diện tích hoặc thể tích LA và SCD. Tuy nhiên, các nghiên cứu sâu hơn sử dụng các phương pháp chính xác hơn để đo LA với mục đích điều tra mối liên quan độc lập với SCD được đảm bảo.
- NSVT
Nhịp nhanh thất tạm thời – được định nghĩa khi ≥3 nhịp thất liên tiếp với tần số ≥120 nhịp mỗi phút và thời gian <30 giây khi theo dõi Holter (thời gian tối thiểu 24 giờ) tại hoặc trước khi đánh giá – là một yếu tố tiên đoán độc lập và được chứng minh rõ bằng tư liệu về SCD (O’Mahony, 2014., Elliott, 2008., Dimitrow, 2010., Monserrat, 2003.)
Tỷ lệ 20-30% bệnh nhân HCM có NSVT được ghi nhận trên Holter ECG. Ngược lại với các yếu tố nguy cơ đã đề cập trước đây, NSVT là một biến nhị phân. Nói cách khác, chỉ sự hiện diện của nhịp nhanh thất bất kể tần suất, thời gian hoặc tỷ lệ NSVT là một yếu tố nguy cơ của SCD. 9. Đáp ứng huyết áp bất thường khi tập thể dục
Khoảng một phần ba bệnh nhân người lớn mắc HCM có phản ứng huyết áp tâm thu bất thường khi gắng sức, đặc trưng bởi hạ huyết áp tiến triển hoặc không làm tăng huyết áp tâm thu do giảm sức cản mạch hệ thống không phù hợp và dự trữ cung lượng tim thấp (Elliott, 2014). Định nghĩa được sử dụng nhiều nhất về đáp ứng huyết áp bất thường là theo trình tự: áp lực suy giảm ≥ 20 mmHg từ khi nghỉ ngơi đến khi gắng sức cao điểm hoặc giảm > 20 mmHg so với huyết áp cao nhất. Một nghiên cứu đơn trung tâm rất cũ đã ghi lại mối liên quan giữa đáp ứng huyết áp bất thường khi gắng sức và nguy cơ cao mắc SCD ở bệnh nhân ≤ 40 tuổi; tuy nhiên, thuần tập này chỉ bao gồm 161 bệnh nhân và điều tra 12 SCD (3 trong nhóm đáp ứng huyết áp bình thường so với 9 SCD trong nhóm đáp ứng huyết áp bất thường) tất cả các bệnh nhân đều có các yếu tố nguy cơ chính khác như: ngất không rõ nguyên nhân, gia đình tiền sử SCD, tắc nghẽn LVOT, NSVT và tất cả các yếu tố nguy cơ này có liên quan đến tuổi trẻ (Sadoul, 1997.). Tóm lại, nhóm thuần tập này có thể có sự sai lệch và có thể nếu chúng ta có khả năng tính điểm HCM nguy cơ đột tử, nó sẽ dẫn đến nguy cơ 5 năm ≥ 6% không phân biệt đối với đáp ứng huyết áp.
Cuối cùng, một nghiên cứu gần đây trên 426 bệnh nhân HCM chỉ có thể liên hệ đáp ứng huyết áp bất thường với các kết quả bất lợi (điểm cuối tổng hợp của tử vong nói chung, phóng điện của máy khử rung tim phù hợp, nhập viện do suy tim) nhưng không chuyên biệt với SCD (Desai, Năm 2014.).
Do đó, tài liệu rõ ràng về mối liên quan độc lập có ý nghĩa thống kê giữa đáp ứng huyết áp bất thường và SCD chưa có trong tài liệu
* Các yếu tố nguy cơ sửa đổi mới và các giải quyết mới
Trong vài năm qua, nghiên cứu tim mạch đang tiến rất nhanh trong việc phát hiện ra các yếu tố nguy cơ điều chỉnh mới. Do đó, chứng phình mỏm LV, đa đột biến gen sarcomere, xơ hóa cơ tim (MF) và giai đoạn cuối (ES) HCM ngày càng trở nên quan trọng hơn với vai trò là phán xử cuối cùng, đặc biệt trong các trường hợp có nguy cơ trung gian.
- Phình mỏm thất trái
Phình mỏm thất trái (LV) được coi là một biến chứng hiếm gặp của HCM, một nghiên cứu báo cáo tỷ lệ hiện mắc là 2,2% trong dân số HCM nói chung (Maron, 2008.). Mặt khác, điều quan trọng cần lưu ý là nghiên cứu này báo cáo tỷ lệ biến cố tim mạch bất lợi là 10,5% mỗi năm (SCD, sốc ICD thích hợp, đột quỵ huyết khối tắc mạch không tử vong và suy tim tiến triển) cao hơn rất nhiều so với của dân số HCM nói chung. Đặc biệt trong loạt bài lớn này, 12 bệnh nhân (43% bệnh nhân phình mỏm LV) gặp biến cố lâm sàng bất lợi bao gồm: 2 SCD, 2 ngừng tim được thoát (1 bệnh nhân có biểu hiện rung thất), được can thiệp ICD thích hợp cho nhịp nhanh thất / rung thất (3 bệnh nhân) và suy tim tiến triển. Điều rất quan trọng cần lưu ý là sáu trong số 12 bệnh nhân này cũng phát triển ES-HCM (rối loạn chức năng tâm thu với phân suất tống máu LV <50%) cho thấy rằng biến chứng này có liên quan đến sự tiến triển của ES và ủng hộ quan điểm của mỏm LV như gót chân Achille trong HCM. Hơn nữa, hầu hết các túi phình có một vành sẹo, có liên quan đến các vùng rộng rãi của MF. Tương tự như vậy, một nghiên cứu rất gần đây cho thấy một gradient từ đáy đến mỏm rõ ràng trong tim ES-HCM với sự gia tăng đáng kể lượng MF từ đáy về phía đỉnh, nơi bị ảnh hưởng nghiêm trọng do xơ hóa (Galati, 2016).
Do đó, nghiên cứu này đã cung cấp một bằng chứng về mối liên hệ giữa Phình mạch Apical LV và ESHCM, có thể liên quan đến MF (Galati, 2016.). Tuy nhiên, cơ chế gây ra sự hình thành của chứng phình động mạch đỉnh ở HCM vẫn chưa được hiểu rõ.
Điều quan trọng cần làm nổi bật là độ dốc từ đáy đến đỉnh này là một đặc điểm khác biệt của sarcomeric ES-HCM, đặc biệt là khi so sánh với các bệnh cơ tim khác có kiểu hình phì đại – đặc biệt là bệnh amyloidosis – nơi mỏm LV tương đối bị loại bỏ bởi sự thâm nhập amyloid (Quarta, 2014.). Tốc độ căng được đánh giá bằng siêu âm tim theo dõi đốm có thể phát hiện đặc điểm này và cung cấp manh mối hữu ích để chẩn đoán phân biệt (Quarta, 2014.).
- Đa đột biến gen Sarcomere
Trừ khi, trong những thập kỷ qua, nhiều nghiên cứu nhỏ đã được công bố về mối liên quan giữa một số “đột biến ác tính” và kết quả bất lợi, những dữ liệu này không được xác nhận sau đó từ các nghiên cứu khác. Mặt khác, trong 5 năm qua, nhiều tiến bộ và bước tiến đã được thực hiện bằng cách sử dụng các kỹ thuật mới về giải trình tự DNA thế hệ tiếp theo và thực hiện các nghiên cứu quốc tế đa trung tâm mới.
Bảy năm trước, một nghiên cứu đã ghi nhận bệnh nhân HCM có đột biến kép hoặc ba có tiên lượng xấu hơn so với những bệnh nhân có đột biến đơn lẻ (Girolami, 2010.). Girolami và cộng sự. đã có thể chứng minh trong một nhóm lớn gồm 488 nhóm tham gia với HCM sự hiện diện của đột biến gen ba sarcomere hiếm gặp có tương quan với sự tiến triển suy tim, SCD và ES-HCM.
Vì vậy, nghiên cứu này đã cung cấp một xác nhận mạnh mẽ của các nghiên cứu trước đó trong HCM tồn tại một “gánh nặng di truyền” và điều này đóng một vai trò đáng kể trong sự biểu hiện kiểu hình của HCM và trong việc sửa đổi bệnh sử tự nhiên của HCM (Girolami, 2010., Richard, 2003., Ingles, 2005., Maron, 2012.). Những bệnh nhân này trẻ hơn và bị xơ hóa cơ tim nhiều hơn và suy giảm nhiều hơn chức năng vi mạch do tái tạo bất lợi của tiểu động mạch vành.
Một nghiên cứu quốc tế đa trung tâm khác gần đây đã ghi nhận tỷ lệ phổ biến kiểu gen phức tạp (đặc trưng bằng sự cùng tồn tại của 2 hoặc 3 đột biến) là 13% trong một nhóm thuần tập gồm 150 bệnh nhân ES-HCM, tức là, một tỷ lệ phần trăm cao gấp ba lần so với các giá trị được báo cáo trong nhóm cũ trước đó tài liệu cho bệnh nhân HCM (Biagini, 2014). Nghiên cứu này củng cố giả thuyết “gánh nặng di truyền” góp phần vào mức độ nghiêm trọng của bệnh và dự đoán sự tiến hóa của ES.
Năm 2010 Ho C và cộng sự. (Ho, 2010.) đã cung cấp chứng minh đầu tiên ở người xơ hóa cơ tim phát triển không chỉ ở những bệnh nhân có biểu hiện HCM rõ ràng và với LGE ở CMR, mà còn ở những người mang đột biến sarcomeric không phì đại thất trái. Họ đã có thể chứng minh – định lượng nồng độ serum C-terminal propeptide of type I procollagen (PICP) trong huyết thanh của procollagen loại I (PICP) cho thấy tăng tổng hợp collagen cơ tim – xơ hóa cơ tim thậm chí còn có trước phì đại thất trái.
Ba năm sau, cùng một nhóm nghiên cứu đã xác nhận quan sát này, so sánh các kỹ thuật CMR khác nhau về đặc điểm mô, tức là phương pháp lập bản đồ T1 so với LGE (Ho, 2013). Trong nghiên cứu này, phương pháp lập bản đồ T1 (pre-và postgadolinium) là kỹ thuật duy nhất có thể xác định xơ hóa mô kẽ không chỉ ở bệnh nhân HCM và những người mang đột biến sarcomere có phì đại thất trái, mà còn ở những người mang đột biến sarcomere không phì đại thất trái, ngược lại LGE chỉ có thể xác định xơ hóa thay thế ở bệnh nhân HCM. Cả bản đồ T1 và LGE đều mở rộng hơn đáng kể trong sarcomeric HCM. Những phát hiện này ghi nhận rằng gánh nặng xơ hóa cao hơn ở những bệnh nhân HCM có đột biến sarcomere so với những người không có đột biến sarcomere và phần nào có thể làm cơ sở cho các kết quả xấu hơn được báo cáo trong HCM sarcomere dương tính so với sarcomere âm tính.
Do đó, những kết quả này có thể cung cấp mối liên hệ giữa sự phát triển xơ hóa cơ tim ở bệnh nhân trẻ hơn và các biến cố SCD liên quan đến tổng lượng xơ hóa cơ tim.
Cuối cùng, vào năm 2015, Lopes và cộng sự cho thấy trong một loạt lớn (gần 900 bệnh nhân HCM) bằng chứng đầu tiên cho thấy bệnh nhân có đột biến gen protein sarcomeric có tỷ lệ tử vong liên quan đến tim mạch và SCD cao hơn trong suốt cuộc đời so với bệnh nhân HCM không có đột biến gen protein sarcomeric. Tuy nhiên, điều quan trọng cần lưu ý là số lượng biến cố kết cục thấp trong quá trình theo dõi có thể làm sai lệch phân tích tỷ lệ sống sót trong nghiên cứu này.
Tóm lại, hiện nay, không có bằng chứng rõ ràng về mối liên hệ giữa một số đột biến cụ thể và SCD ở bệnh nhân HCM và chính xác là không thể đưa biến này vào đánh giá nguy cơ SCD. Do đó, nhiều nghiên cứu tiền cứu quốc tế đa trung tâm hơn nữa được đảm bảo.
Mặc dù vậy, cần phải tính đến “gánh nặng di truyền” liên quan đến kết quả bất lợi ở bệnh nhân HCM, đặc biệt ở bệnh nhân trẻ tuổi. Thật vậy, những bệnh nhân có đột biến kép hoặc đột biến ba này cần được theo dõi rất nghiêm ngặt và đánh giá sớm để cấy ICD, vì đây là yếu tố thay đổi mạnh mẽ tiền sử tự nhiên của HCM.
- Xơ cơ tim
Trong những năm gần đây, một số nghiên cứu cơ bản và nghiên cứu lâm sàng có liên quan đã tìm hiểu ý nghĩa của xơ cơ tim (Myocardial fibrosis: MF) trong HCM. Cả loại xơ hóa mô kẽ thay thế và lan tỏa đều được mô tả là dấu hiệu mô học của bệnh. Trong khi quá trình xơ hóa thay thế đã xảy ra từ lâu ở HCM, đặc biệt là ở giai đoạn cuối của bệnh cơ tim phì đại (ES-HCM) (Moon, 2004.), gần đây người ta mới chỉ ra sự lắng đọng collagen kẽ có thể xảy ra trong giai đoạn rất sớm của HCM và thậm chí trước khi phát triển phì đại. Thật vậy, xơ hóa đại diện cho một trong những biểu hiện kiểu hình chính của HCM và không nhất thiết là một biến chứng liên quan đến thời gian (Ho, 2010., Ho, 2013.). Nguyên bào sợi và nguyên bào sợi cơ đã được xác định là những tác nhân gây xơ hóa chính trong cơ tim và do đó chịu trách nhiệm tổng hợp các protein nền ngoại bào. Một vai trò quan trọng đối với tăng sinh nguyên bào sợi trong bệnh xơ hóa đã được gợi ý liên quan đến HCM; đặc biệt, việc truyền tín hiệu bằng cách biến đổi yếu tố tăng trưởng β (TGF β) dường như rất quan trọng đối với việc kích hoạt các nguyên bào sợi. Ở những bệnh nhân bị HCM, sự tích tụ collagen ngày càng tăng dẫn đến sự xơ hóa có thể xuất hiện dưới dạng tế bào mô kẽ lan tỏa, dày đặc và thay thế (“giống sẹo”) hoặc loại hỗn hợp. Các nghiên cứu trước đây trên mô hình chuột đã ghi lại rằng tốc độ tăng sinh của nguyên bào sợi, xảy ra độc lập với sự tăng sinh của tế bào sợi, tăng liên tục trong tim của những con chuột mang đột biến trong myosin 7 (Frey, 2011., Teekakirikul, 2010.). Hơn nữa, như đã viết trước đây (xem đoạn “Nhiều đột biến gen Sarcomere”) Người mang đột biến gen sarcomere biểu hiện kiểu hình thúc đẩy xơ (profibrotic) trở nên rõ ràng trước khi biểu hiện phì đại và những đối tượng này có gánh nặng xơ sợi lớn hơn những đối tượng không có đột biến gen sarcomeric. không phụ thuộc vào sự tăng sinh của tế bào, được tăng liên tục trong tim của những con chuột mang đột biến trong myosin 7 (Frey, 2011., Teekakirikul, 2010.). Hơn nữa, như đã được trình bày trước đây (xem đoạn “Nhiều đột biến gen Sarcomere”) Người mang đột biến gen sarcomere biểu hiện kiểu hình thúc đẩy xơ trở nên rõ ràng trước khi biểu hiện phì đại và những đối tượng này có gánh nặng xơ sợi lớn hơn những đối tượng không có đột biến gen sarcomeric.
Tuy nhiên, người ta vẫn chưa hiểu rõ mối liên hệ chính xác giữa đột biến sarcomeric và tăng sinh tế bào non.
Sẹo xơ trong tim tương quan chặt chẽ với việc gia tăng tần suất loạn nhịp tim và SCD.
Phần lớn dữ liệu về MF ở HCM được lấy từ các nghiên cứu CMR (Ho, 2013., Bruder, 2010., O’Hanlon, 2010., Adabag, 2008., Maron, 2008., Rubinshtein, 2010., Todiere, 2012, Prinz, 2013., Chan, 2014., Ismail, 2014., Green, 2012., Briasoulis, 2015.) hoặc gián tiếp từ nghiên cứu về các dấu ấn sinh học chu chuyển collagen huyết thanh như MMP-1 (matrix metalloproteinase-1) , PICP (telopeptide tận cùng C của collagen loại I), và TIMP-1 (chất ức chế mô của metalloproteinase-1) (Ho, 2010.).
Một nghiên cứu gần đây đã cung cấp đặc điểm mô học và mô học tốt nhất của MF ở ES-HCM và cung cấp sự so sánh chính xác nhất giữa LGE-CMR và định lượng mô học của MF (xem phần ES-HCM) (Ho, 2010).
Hơn nữa, một số lượng lớn các nghiên cứu về CMR (Bruder, 2010., O’Hanlon, 2010., Adabag, 2008., Maron, 2008., Rubinshtein, 2010., Todiere, 2012., Prinz, 2013., Chan, 2014. , Ismail, 2014.) và hai phân tích tổng hợp (Green, 2012., Briasoulis, 2015.) về vai trò tiên lượng của MF đã được công bố và người ta đã ghi nhận rằng MF chắc chắn là một yếu tố dự báo nguy cơ mạnh độc lập về tử vong tim mạch và SCD ở HCM. Đặc biệt, một nghiên cứu gần đây cho thấy mức độ LGE ≥15% khối lượng LV làm tăng nguy cơ SCD > 2 lần và lượng LGE ≥20% làm tăng > 3 lần nguy cơ tiến hóa ES-HCM (Chan, Năm 2014.).
Mặc dù vai trò tiên lượng được ghi nhận rõ ràng của MF ở HCM (phân tích lần cuối xem xét 1414 bệnh nhân không có LGE và 1653 bệnh nhân có LGE với thời gian theo dõi trung bình là 3,05 năm) (Briasoulis, 2015), hiện tại cũng không phải hướng dẫn của châu Âu cũng không phải hướng dẫn của Mỹ đưa nó vào yếu tố nguy cơ SCD. Tuy nhiên, điều quan trọng cần nhấn mạnh là các hướng dẫn của Mỹ thừa nhận MF là một quyết định tiềm năng, trong khi các hướng dẫn của Châu Âu không đưa nó vào mô hình tiềm năng.
Đây là một nghịch lý nếu chúng ta nhận thấy rằng MF đã được nghiên cứu rộng rãi trong mười năm qua và có nhiều nghiên cứu và hai phân tích tổng hợp về vai trò tiên lượng của nó. Do đó, vai trò tiên lượng của MF ở HCM thậm chí còn dựa trên bằng chứng tốt hơn so với một số yếu tố nguy cơ bệnh sử kinh điển.
- Bệnh cơ tim phì đại gia đoạn cuối
Giai đoạn cuối của bệnh cơ tim phì đại (ES-HCM, hoặc thoái hóa của bệnh cơ tim phì đại giảm động – giãn) (Hình 3) là một biến chứng hiếm gặp của HCM (5% tổng số bệnh nhân bị HCM) được xác định là phân suất tống máu LV ≤ 50% khi nghỉ. , phản ánh rối loạn chức năng tâm thu toàn thể và mang một tiên lượng đáng ngại (Harris, 2006., Biagini, 2005., Thaman, 2005., Biagini, 2008., Melacini, 2010.). Tỷ lệ tử vong nói chung là 11% mỗi năm, trái ngược hẳn với tỷ lệ 1-2% mỗi năm của dân số HCM nói chung, và là do suy tim trơ và SCD. ES-HCM gần như là chỉ định ghép tim duy nhất ở tất cả các trung tâm trên thế giới cho bệnh nhân bị ảnh hưởng của HCM (Harris, 2006., Biagini, 2005., Thaman, 2005., Biagini, 2008., Melacini, 2010.).
Người ta vẫn còn tranh luận về việc liệu ES-HCM có đại diện cho một chỉ định để cấy ICD hay không.
Một số cơ chế khác nhau đã được đề xuất để giải thích sự tiến triển thoái hóa đối với ES, nhưng cơ chế chính là hai cơ chế: xơ hóa cơ tim và thiếu máu cục bộ vi mạch. Người ta cũng đưa ra giả thuyết rằng sự phát triển của ES-HCM có thể bị ảnh hưởng bởi nền tảng di truyền cụ thể (nhiều đột biến gen sarcomere)
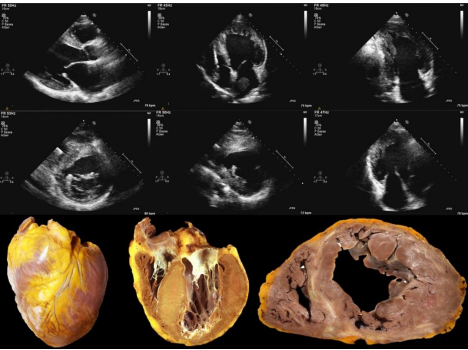
Hình 3. Đặc tính siêu âm tim và sinh bệnh học của bệnh cơ tim phì đại giai đoạn cuối (tiến triển thoái hóa giãn – giảm động).
Bệnh cơ tim phì đại giai đoạn cuối được đặc trưng bằng giãn và giảm động thất trái với thành thất trái mỏng đi hoặc bình thường. Các hình ảnh siêu âm cho thấy (trong trường hợp này) có các thrombus trong nhĩ trái. Các hình ảnh bệnh lý chỉ ra xơ hóa là cơ tim bệnh trong các đường trắng.
- Gần đây, một nghiên cứu đa trung tâm đầu tiên của châu Âu đã cung cấp đánh giá mô học định lượng chi tiết đầu tiên về mức độ, sự phân bố, các mô hình và các loại MF trong một quần thể lớn các bệnh tim mạch ES-HCM. Hơn nữa, nó đánh giá mối quan hệ định lượng giữa đánh giá LGE-CMR và phân tích mô học của MF. Nó khám phá những quan điểm mới về MF, về đánh giá hình ảnh và cuối cùng là về sinh lý bệnh của HCM và SCD (Galati, 2016.).
Trước đây, các đánh giá mô học chi tiết và hệ thống về các đặc điểm định lượng và định tính của MF ở HCM còn thiếu (chỉ có các nghiên cứu mô học giới hạn trong sinh thiết đơn lẻ hoặc các mẫu vách trước-đáy thu được trong quá trình phẫu thuật nội soi). Hơn nữa, các nghiên cứu về mối tương quan giữa mô học và CMR rất ít và chỉ giới hạn ở các mối tương quan định tính.
Lần đầu tiên nghiên cứu đa trung tâm này đã cung cấp một số hiểu biết mới tuyệt vời về nhóm bệnh nhân này, tức là
- Hơn một phần ba cơ tim LV (giá trị trung bình là 37%) được thay thế bằng xơ hóa, do đó tổng lượng MF trong toàn bộ tim này rất cao;
- Độ chệnh mỏm đến đáy rõ ràng xuất hiện với sự gia tăng đáng kể lượng MF từ đáy về phía đỉnh, luôn bị ảnh hưởng nghiêm trọng do MF trong ES-HCM;
- Các thành LV dưới, trước và trước bên cũng như vách liên thất bị ảnh hưởng tối đa bởi sự xơ hóa, trong khi vách LV bên dưới luôn không có một cách tương đối;
- Lớp giữa thành là lớp bị ảnh hưởng nhiều nhất đối với lớp dưới màng tim, lớp dưới cơ tim và lớp màng ngoài. Hơn nữa, mô hình chủ yếu là thành giữa, tiếp theo là thành giữa và dưới màng tim, xuyên màng cứng và giữa thành giữa và dưới cơ tim. Lớp dưới cơ tim và màng dưới tim có thể liên quan nhưng không bao giờ là duy nhất (Hình 4);
- Trong tim ES-HCM có ít nhất ba loại xơ hóa định tính, tức là thay thế (hoặc giống như sẹo), tế bào mô kẽ-quanh cơ và xơ hóa hỗn hợp. Xơ hóa thay thế là loại được biểu hiện nhiều nhất (Hình 5);
- Bệnh nhân trẻ hơn bị xơ hóa hỗn hợp, cả mô kẽ và sẹo, bệnh nhân lớn tuổi chỉ có xơ hóa dạng sẹo là điển hình của giai đoạn tiến triển hơn của bệnh;
- Có sự gần gũi giữa các vùng xơ hóa giống sẹo và các tiểu động mạch bất thường ở các phần khác nhau. Hơn nữa, sự phì đại của các chất trung gian và sự tăng sản của các cơ tạo ra rối loạn chức năng vi mạch vành và là nguyên nhân tiềm ẩn của thiếu máu cục bộ cơ tim;
- Có sự chồng chéo tốt giữa LGE-CMR và định lượng mô học của MF. Tuy nhiên, LGE đã đánh giá thấp mức độ xơ hóa ở những bệnh nhân có MF chủ yếu là tế bào mô kẽ-quanh cơ, cho thấy LGE chỉ có thể xác định được các vết sẹo dày đặc nhưng không thể nắm bắt được sự mở rộng lan tỏa hơn của không gian ngoại bào, chẳng hạn như do xơ hóa mô kẽ.
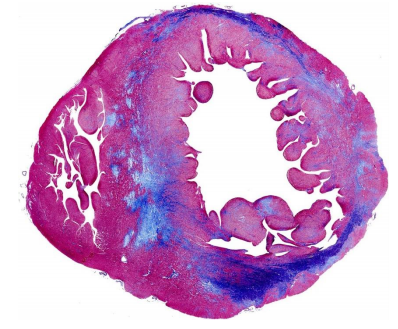
Hình 4. Sự phân bố xơ hóa cơ tim trên tim ES-HCM được bộc lộ ở mặt cắt giữa thất.
Nhuộm ba màu Azan-Mallory. Xơ hóa có màu xanh lam, trong khi cơ tim có màu hồng / đỏ. Lưu ý sự phân bố theo chu vi của xơ hóa chủ yếu là thành giữa trong trường hợp này. Dưới nội và ngoại tâm mạc có thể có liên quan, nhưng không bao giờ là duy nhất (Từ Galati và cộng sự) (Galati 2016).
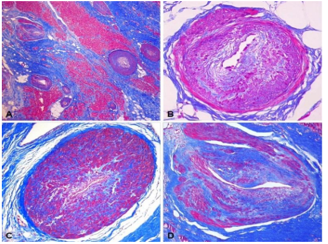
Hình 5. Các type khác nhau của xơ cơ tim trong bệnh cơ tim phì đại giai đoạn cuối (Theo Galati và cộng sự) (Galati 2016).
A: type xơ hóa liên kết quanh tế bào cơ. B: type xơ hóa hỗn hợp. C: type xơ hóa thay thế (như sẹo).
Tóm lại, bệnh nhân ES-HCM có mức độ LGE> 20% khối lượng LV và điều này cộng ang vào hiện tượng thiếu máu cục bộ cơ tim cung cấp giải thích về tỷ lệ tử vong ang năm cao do cả SCD và suy tim trơ.
Cuối cùng, điều quan trọng cần lưu ý là thiếu các nghiên cứu tiền cứu ở bệnh nhân ES-HCM, nhưng sự tiến triển thoái hóa của ES hiện nay khá khó đoán (ES-HCM và SCD có yếu tố gia đình là những yếu tố nguy cơ mạnh nhất), và thường là rất nhanh – thời gian trung bình từ việc xác định ES cho đến tử vong hoặc ghép tim thường là ba năm trong tất cả các bài đã được tìm thấy trong tư liệu đã được xuất bản – do đó rất khó để thiết kế một nghiên cứu tiền cứu. Vấn đề quy mô mẫu nhỏ là do sự hiếm có của quá trình tiến triển thoái hóa ES đối với tổng dân số bị ảnh hưởng do HCM và các nghiên cứu lớn hơn về loại này khó có thể thu lượm được.
Tuy nhiên, các trung tâm chuyển tuyến đại học giải quyết những bệnh nhân này thường chỉ định cấy ICD trong dự phòng tiên phát, vì bệnh nhân ES-HCM còn rất trẻ và thường trong vòng 3 năm họ sẽ được ghép tim. Do đó, nguy cơ bị viêm nội tâm mạc nhiễm trùng ở những bệnh nhân này là rất thấp và lợi ích tiềm tàng là rất cao hoặc không thể lường trước được (tức là thoát khỏi SCD khi bệnh nhân đang trong danh sách chờ ghép tim).
Một điểm khác đáng được đề cập là vai trò của liệu pháp tái đồng bộ tim (CRT) đối với nhóm bệnh nhân này. Một nghiên cứu hồi cứu gần đây, được công bố vào năm 2016 và được thực hiện tại Hoa Kỳ (Killu, 2016.), cho thấy không có lợi ích ở bệnh nhân CRT-D so với nhóm chứng. Không có bất kỳ lợi ích nào về kết quả và CRT-D cũng không thể làm chậm sự tiến triển của bệnh, do đó, giống như ở những bệnh nhân không có CRT-D, thời gian từ chẩn đoán ES-HCM đến ghép tim hoặc cấy thiết bị trợ giúp thất trái là ba năm. Ngay cả phân suất tống máu thất trái vẫn giữ nguyên trừ khi các thông số điện sinh lý đối với cấy CRT-D được tôn trọng (tức là thời gian QRS cơ bản là 182 ± 39 msec, với hình thái bó nhánh trái hoặc phải và không có rung nhĩ trong quần thể được cấy). Điều này có thể được giải thích do số lượng xơ hóa cơ tim rất cao trong cơ tim ở bệnh này, vì vậy không thể điều trị bằng điện để tái đồng bộ hóa các vết sẹo thay thế một tỷ lệ cơ tim có liên quan như vậy.
* Các điểm cần chú ý
Ước tính nguy cơ SCD là một trong những chủ đề khó nhất của toàn bộ tim mạch học. Điều này là do một số yếu tố:
- Dân số HCM trẻ hơn so với phần còn lại của dân số tim mạch, do đó quyết định cấy hay không cấy ICD có ảnh hưởng lớn đến cuộc sống của những bệnh nhân này và chỉ một sai lầm có thể là thảm họa.
- Việc quản lý bệnh nhân bị ảnh hưởng do HCM đòi hỏi một đánh giá toàn diện, tỉ mỉ và phù hợp mà không thể được thực hiện bằng một bác sĩ tim mạch hoặc một bác sĩ điện sinh lý duy nhất ở bất kỳ trung tâm nào mà phải do một nhóm bác sĩ tim mạch có chuyên môn đã được tôi luyện về các bệnh cơ tim và màng ngoài tim. Người làm việc trong các trung tâm giới thiệu cho HCM.
- Việc đánh giá nguy cơ rất phức tạp do thiếu các nghiên cứu tiền cứu.
- Thật không may, sự đồng thuận quốc tế về đánh giá nguy cơ SCD vẫn còn thiếu đến cả thế giới về lập luận này gần như bị chia rẽ theo hai quan điểm: quan điểm của Mỹ và châu Âu.
- Tính đến tất cả những khó khăn này và nỗ lực và phải thực hiện một bước tiến. Một trong những điểm cốt yếu là việc giải thích chính xác các yếu tố nguy cơ bệnh sử dưới ánh sáng của bằng chứng đương đại và việc thực hiện các yếu tố nguy cơ mới không thể bỏ qua. Vì vậy, chia sẻ vị trí của người Mỹ hay người Châu Âu không quan trọng, vì không ai là chủ sở hữu của sự thật. Chúng ta cần đưa ra các quyết định và quản lý bệnh nhân HCM trên cơ sở các bằng chứng rõ ràng nhất. Do đó, hiện tại các tài liệu cho thấy mô hình châu Âu chính xác hơn và nó đánh giá nguy cơ SCD tốt hơn so với mô hình cũ của Mỹ.
- Mặt khác, các yếu tố nguy cơ sửa đổi mới có tầm quan trọng đáng kinh ngạc và các chuyên gia Mỹ nhận thức rõ hơn về những yếu tố này so với các chuyên gia châu Âu. Vì vậy, đại đa số các trung tâm chuyển tuyến HCM coi tổng lượng LGE và xơ hóa cơ tim là một yếu tố nguy cơ điều chỉnh quan trọng có thể ảnh hưởng đến quyết định cấy hay không cấy ICD.
- Các trung tâm dành riêng cho HCM trên toàn thế giới nên hợp tác và thực hiện các nghiên cứu tiền cứu quốc tế đa trung tâm với mục đích khám phá các yếu tố dự báo đáng tin cậy về SCD ở HCM và đưa ra các quyết định dựa trên bằng chứng tốt hơn.
- Sự cần thiết của một thỏa thuận quốc tế là một vấn đề hấp dẫn không thể trì hoãn thêm. Vì vậy, mục đích của chương này không chỉ là xem xét cách quản lý cũ đối với nguy cơ SCD ở bệnh nhân HCM, mà còn đặc biệt tập trung vào các yếu tố nguy cơ sửa đổi mới và các quan điểm trong tương lai. Sau đó, mục đích là nhấn mạnh có một công việc và nghiên cứu tuyệt vời phải làm trong lĩnh vực này và nó không thể được thực hiện bằng một trung tâm duy nhất hoặc bằng một quốc gia duy nhất (bởi vì trong khoa học và y học không ai đến trước, do đó “không phải nước Mỹ trước tiên”, “Không phải châu Âu trước tiên”, mà là “thế giới đến trước”.
* Triển vọng tương lai
Một nỗ lực phi thường cần được thực hiện trong việc tìm hiểu về bệnh xơ hóa cơ tim và mối liên hệ giữa di truyền, xơ hóa cơ tim và thiếu máu cục bộ cơ tim.
Nhiều nghiên cứu đã được công bố đã nêu bật tầm quan trọng đáng chú ý của xơ hóa về cơ chế bệnh sinh của sự tiến hóa HCM, SCD và ES-HCM.
- Nghiên cứu về bệnh xơ hóa cơ tim
Một trong những giả thuyết có liên quan nhất được xác nhận bằng một số nghiên cứu (cả trong mô hình ở chó và ở người) là các đột biến gây bệnh cụ thể không chỉ của các gen sarcomere mà còn của các gen mã hóa cho các protein nền ngoại bào – và điều chỉnh sự tăng sinh nguyên bào sợi và nguyên bào sợi cơ – kích thích cả hai tổng hợp collagen, tăng sinh nguyên bào sợi (gây ra quá trình tái cấu trúc tim tiền nguyên bào sớm trước phì đại thất trái) và rối loạn chức năng vi mạch vành được đặc trưng bằng tăng sản nội mạc và phì đại trung gian dẫn đến giảm và đôi khi gây tắc nghẽn lòng mạch (Hình 6). Những đặc điểm này dễ thấy hơn ở những bệnh nhân trẻ tuổi và ở ES-HCM, cũng như được ghi lại bằng một nghiên cứu được trích dẫn.
Theo luận điểm này (Hình 7), trong giai đoạn đầu quá trình xơ hóa phát triển như xơ hóa tế bào mô kẽ và chỉ có thể được phát hiện bằng phương pháp CMR ánh xạ T1 mới hoặc bằng cách định lượng các dấu ấn sinh học chu chuyển collagen cụ thể (như PICP, v.v.). Bệnh nặng dần và xơ hóa mô kẽ hội tụ ở những vùng sẹo dày đặc hoặc xơ hóa thay thế. Khi tổng lượng xơ hóa đạt đến một mức độ nhất định và một ngưỡng tới hạn (thường là ≥15% khối lượng LV), các biến cố loạn nhịp chính gây ra SCD sẽ phát sinh.
Một nỗ lực phi thường cần được thực hiện trong việc tìm hiểu về bệnh xơ hóa cơ tim và mối liên hệ giữa di truyền, xơ hóa cơ tim và thiếu máu cục bộ cơ tim.
Nhiều nghiên cứu đã được trích dẫn trước đây trong chương này đã nêu bật tầm quan trọng đáng chú ý của xơ hóa về cơ chế bệnh sinh của sự tiến triển thoái hóa HCM, SCD và ES-HCM.
Hình 6. Các động mạch vành trong cơ bất thường kết hợp với xơ hóa thay thế (theo Galati và cộng sự.) (Galati 2016)
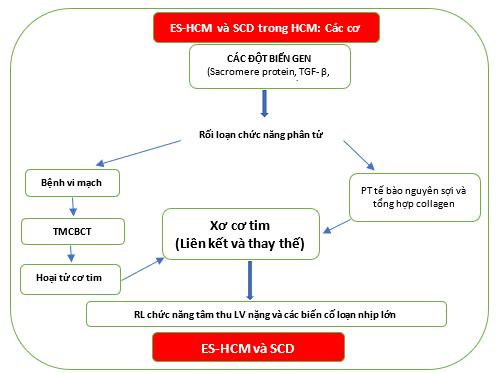
Hình 7. Các cơ chế sinh lý bệnh và tiến triển thoái hóa của ES-HCM ở bệnh nhân HCM.
ES-HCM: bệnh cơ tim phì đại giai đoạn cuối. SCD: đột tử tim. TMCTCB: thiếu máu cơ tim cục bộ. RL: rối loạn. LV: thất trái.
Hơn nữa, sự tiến triển thêm của xơ hóa – đặc biệt là các đối tượng có khuynh hướng di truyền – và các bất thường vi mạch vành gây ra sự tiến triển đáng ngại nhất, tức là sự tiến triển thoái hóa của ES-HCM. TGFβ đóng một vai trò quan trọng trong quá trình này.
Một điểm quan trọng khác cần nhấn mạnh là một số lượng lớn các nghiên cứu cho thấy MF ban đầu ảnh hưởng đến lớp giữa của thành và sau đó – tùy thuộc vào cơ chế bệnh sinh phổ biến (thiếu máu cục bộ cơ tim, sản xuất collagen và tăng sinh nguyên bào sợi) – lan tỏa ở dưới thượng tâm mạc đôi khi ở dưới nội tâm mạc và ở những trường hợp gia tăng trở nên xuyên thành. Đặc điểm này giúp phân biệt rõ ràng sarcomeric ES-HCM với bệnh tim thiếu máu cục bộ – nơi xơ hóa chủ yếu ở dưới cơ tim và lan tràn từ dưới nội tâm mạc về phía dưới thượng tâm mạc – và với bệnh cơ tim loạn nhịp trong đó xơ hóa chủ yếu là dưới thượng tâm mạch với sự tiến triển dưới thượng-dưới nội tâm mạch (Galati, 2016). Người ta vẫn chưa hiểu được lý do của sự tham gia chính của lớp giữa thành tim cũng như phần mỏm đối với phần nền. Do đó, nghiên cứu trong lĩnh vực này và sự hiểu biết về các cơ chế phân tử là cơ sở của quá trình sản xuất xơ hóa quá mức và bệnh lý có thể giúp khoa học tim mạch khám phá ra các liệu pháp cụ thể mới, nhằm mục đích ngăn chặn hoặc giảm sự phát triển xơ hóa. Một ví dụ rõ ràng có thể là sự phát triển của các kháng thể đơn dòng mới ngăn chặn TGF-β hoặc các protein và gen liên quan đến quá trình tạo xơ.
- NGHIÊN CỨU VỀ THIẾU MÁU CỤC BỘ CƠ TIM VÀ RỐI LOẠN CHỨC NĂNG VI MẠCH
Bắt đầu từ các nghiên cứu sau khi khám nghiệm tử thi trên một loạt nhỏ bệnh nhân đột tử, trong thập kỷ qua, các nghiên cứu khám nghiệm tử thi ở bệnh nhân HCM đã ghi nhận bệnh lý vi mạch vành có đặc điểm là tăng sinh nội mạc và phì đại lớp giữa gây giảm diện tích lòng mạch của tiểu động mạch vành trong cơ và đôi khi làm tắc.
Năm 2003, Cecchi và cộng sự cung cấp bằng chứng đầu tiên cho thấy thiếu máu cục bộ có hậu quả tiên lượng đáng kể trong HCM. Thật vậy, bằng cách sử dụng NH13-Positron Emission Tomography (PET) với tiêm dipyridamole người ta đã ghi nhận – trong một nhóm thuần tập gồm 51 bệnh nhân HCM được theo dõi trong hơn 8 năm – lưu lượng máu chảy ra từ cơ tim được chứng minh là một yếu tố dự báo độc lập về tử vong tim mạch (do SCD hoặc dẫn đến suy tim trơ) (Cecchi, 2003.).
Thật vậy, các khu vực xơ hóa bắt nguồn từ sự thay thế của các tế bào cơ tim bị hoại tử hoặc chết theo hình thái được coi là dấu hiệu hình thái tiềm ẩn của rối loạn nhịp tim do khởi kích kiểu siêu nhỏ.
Sau các nghiên cứu đa trung tâm trên loạt bệnh nhân HCM lớn hơn (Maron, 2009., Olivotto, 2011., Olivotto, 2006., Petersen, 2007.) xác nhận phát hiện này không chỉ sử dụng PET mà còn cả CMR, đặc biệt là chúng có thể chứng minh mối quan hệ giữa bất thường vi mạch nghiêm trọng, thiếu máu cục bộ cơ tim nghiêm trọng và sự tiến triển của ES-HCM dẫn đến suy tim trơ. (Cecchi, 2003., Olivotto, 2006.).
Hơn nữa, gần đây, người ta đã chứng minh rằng những bệnh nhân có đột biến gen sợi cơ sarcomere – đặc biệt là những người có đột biến MHY7 và những bệnh nhân có đột biến đôi hoặc ba – đối với những bệnh nhân không có đột biến gen sarcomere cho thấy thiếu máu cục bộ vi mạch đáng kể hơn do một bệnh lý của các mạch vành nhỏ nặng hơn (Maron, 2009., Olivotto, 2011.).
Các nghiên cứu CMR đã chứng minh lưu lượng máu cơ tim bị giảm ở mức độ lớn hơn ở phần dưới cơ tim, và vùng thiếu máu cục bộ vi mạch có liên quan đến LGE.
Cuối cùng, một nghiên cứu được đề cập trước đây, đã xác nhận những quan sát này từ quan điểm mô bệnh học, ghi lại mối quan hệ chặt chẽ giữa bệnh lý vi mạch vành chính và sự phát triển của xơ hóa thay thế.
Giả thuyết được công nhận nhiều nhất là các đột biến gây bệnh liên quan đến hệ thống có nguồn gốc tiền thượng tâm mạc như tế bào hình thành mạch vành, van hai lá và khung xơ tim có thể gây ra tất cả các bất thường về mô bệnh học được quan sát thấy ở bệnh nhân HCM (Maron, 2009., Olivotto, 2011., Olivotto, 2006., Petersen, 2007.).
Tuy nhiên, các nghiên cứu sâu hơn cần được đảm bảo trong lĩnh vực này, vì đây là một hiện tượng đáng chú ý khác có thể là một mục tiêu điều trị tiềm năng.
- NGHIÊN CỨU DI TRUYỀN
Ngoài quan điểm tập trung vào tế bào cơ tim về chấn thương tim, hiện nay người ta đã chấp nhận rằng những thay đổi của chất nền ngoại bào tim (ECM) và tái cấu trúc tim đóng một vai trò quan trọng trong sự phát triển và tiến triển của các bệnh tim dẫn đến SCD và suy tim
Trong chương này, chúng tôi thường trích dẫn và chỉ ra vai trò chính của đột biến gen quy định ECM và tăng sinh nguyên bào sợi trong sự phát triển của xơ hóa cơ tim và trên kiểu hình HCM.
Tương tự, gen tương tự có thể gây ra bệnh lý của các tiểu động mạch vành cũng như một số đột biến gen sarcomere cụ thể. Hơn nữa, chúng tôi cũng đề cập đến vai trò của đột biến kép và đột biến ba – tức là khái niệm “gánh nặng di truyền”.
Thêm vào đó, một khía cạnh khác đáng được đề cập. Gần đây, một nghiên cứu đa trung tâm của Ý (Biagini, 2016.) đã ghi nhận mẫu điện tâm đồ “giả STEMI”, thời gian QRS ≥120 msec và khoảng QTc kéo dài là những yếu tố dự báo độc lập của SCD và các biến cố tim mạch lớn (shock của ICD phù hợp, ngừng tim được hồi sức, tử vong do suy tim, đột quỵ tim mạch hoặc ghép tim). Phát hiện sơ bộ này cần được xác nhận bằng các nghiên cứu đa trung tâm quốc tế hơn nữa.
Tuy nhiên, hiện tượng này có thể được giải thích là do sự tồn tại đồng thời của đột biến gen sarcomere và đột biến kênh ion có thể ảnh hưởng đến một nhóm dân số HCM được chọn và có thể dẫn đến SCD đặc biệt sớm.
Thật vậy, điện tâm đồ vẫn là một công cụ có độ nhạy cao trong các bệnh cơ tim và nó có thể phát hiện ra những thay đổi lớn trong cơ tim. Ví dụ, bệnh nhân ES-HCM rất thường xuất hiện “dấu hiệu mũi tên” trên điện tâm đồ (Hình 8), dấu hiệu này thường có thể nhận biết được trước khi chẩn đoán bằng siêu âm tim (Oreto, 2009.)
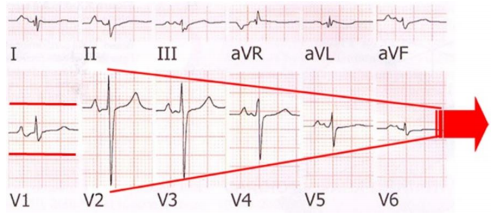
Hình 8. “Dấu hiệu mũi tên”. Các đạo trình trước tim đặt cạnh nhau vẽ một đầu mũi tên. Trong khi V1 đại diện cho trục mũi tên (Theo Oreto) (Oreto 2009).
Cuối cùng, các kỹ thuật giải trình từ thế hệ mới (novel next generation sequencing: NGS) và kiến thức chuyên môn mới ngày càng phát triển cần thiết để hiểu kết quả của kỹ thuật từ thế hệ mới sẽ cho phép các chuyên gia bệnh cơ tim khám phá và hiểu rõ hơn vai trò của di truyền trong sinh lý bệnh trong HCM, đặc biệt là trong sinh lý bệnh SCD.
(Còn nữa)
TÀI LIỆU THAM KHẢO
- Adabag, A. S., Maron, B. J., Appelbaum, E., et al. (2008). Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol, 51, 1369-1374.
- Biagini, E., Coccolo, F., Ferito, M., et al. (2005). Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol, 46, 1543-1550.
- Biagini, E., Olivotto, I., Iascone, M., et al. (2014). Significance of sarcomere gene mutations analysis in the end-stage phase of hypertrophic cardiomyopathy. Am J Cardiol, 114, 769-776.
- Biagini, E., Pazzi, C., Olivotto, I., et al. (2016). Usefulness of electrocardiographic patterns at presentation to predict long-term risk of cardiac death in patients with hypertrophic cardiomyopathy. Am J Cardiol., 118, 432-9.
- Biagini, E., Spirito, P., Leone, O., et al. (2008). Heart Transplantation in Hypertrophic Cardiomyopathy. Am J Cardiol, 101, 387-392.
- Briasoulis, A., Mallikethi-Reddy, S., Palla, M., et al. (2015). Myocardial fibrosis on cardiac magnetic resonance and cardiac outcomes in hypertrophic cardiomyopathy: a meta-analysis. Heart, 101, 1406-11.
- Bruder, O., Wagner, A., Jensen, C. J., et al. (2010). Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol, 56, 875-887.
- Cecchi, F., Maron, B. J. & Epstein, S. E. (1989). Long-term outcome of patients with hypertrophic cardiomyopathy successfully resuscitated after cardiac arrest. J Am Coll Cardiol, 13, 1283-8. Cecchi, F., Olivotto, I., Gistri, R., et al. (2003).
- Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med, 349, 1027-35.
- Chan, R. H., Maron, B. J., Olivotto, I., et al. (2014). Prognostic Value of Quantitative Contrast-Enhanced Cardiovascular Magnetic Resonance for the Evaluation of Sudden Death Risk in Patients With Hypertrophic Cardiomyopathy. Circulation, 130, 484-495.
- Christiaans, I., van Engelen, K., van Langen, I. M., et al. (2010). Risk stratification for sudden cardiac death in hypertrophic cardiomyopathy: systematic review of clinical risk markers. Europace, 12, 313-321.
- Connolly, S. J., Gent, M., Roberts, R. S., et al. (2000). Canadian implantable defibrillator study(CIDS): a randomized trial of the implantable cardioverter defibrillator against amiodarone. Circulation, 101, 1297-1302.
- Connolly, S. J., Hallstrom, A. P., Cappato, R., et al. (2000). Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. AVID, CASH and CIDS studies. Antiarrhythmics vs Implantable Defibrillator study. Cardiac Arrest Study Hamburg. Canadian Implantable Defibrillator Study. Eur Heart J, 21, 2071-2078.
- Desai, M. Y., Bhonsale, A., Patel, P., et al. (2014). Exercise echocardiography in asymptomatic HCM: exercise capacity, and not LV outflow tract gradient predicts long-term outcomes. JACC Cardiovasc Imaging, 7, 26–36.
- Dimitrow, P. P., Chojnowska, L., Rudzinski, T., et al. (2010). Sudden death in hypertrophic cardiomyopathy: old risk factors re-assessed in a new model of maximalized follow-up. Eur Heart J, 31, 3084-3093. Efthimiadis, G. K., Parcharidou, D. G., Giannakoulas, G., et al. (2009).
- Left ventricular outflow tract obstruction as a risk factor for sudden cardiac death in hypertrophic cardiomyopathy. Am J Cardiol, 104, 695-699.
- Elliott, P., Andersson, B., Arbustini, E., et al. (2008). Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J, 29, 270-276.
- Elliott, P. M. (2016). Hypertrophic Cardiomyopathy: Job Done or Work in Progress? J Am Coll Cardiol, 67, 1410-11.
- Elliott, P. M., Anastasakis, A., Borger, M. A., et al. (2014). 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J, 35(39), 2733-79.
- Elliott, P. M., Gimeno Blanes, J. R., Mahon, N. G., et al. (2001). Relation between severity of left-ventricular hypertrophy and prognosis in patients with hypertrophic cardiomyopathy. Lancet, 357, 420-424.
- Elliott, P. M., Gimeno, J. R., Tomé, M. T., et al. (2006). Left ventricular outflow tract obstruction and sudden death risk in patients with hypertrophic cardiomyopathy. Eur Heart J, 27, 1933-1941.
- Elliott, P. M., Poloniecki, J., Dickie, S., et al. (2000). Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol, 36, 2212- 2218.
- Elliott, P. M., Sharma, S., Varnava, A., et al. (1999). Survival after cardiac arrest or sustained ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol, 33, 1596-601.
- Frey, N., Luedde, M. & Katus, H. A. (2011). Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol, 9, 91-100 Galati, G., Leone, O., Pasquale, F., et al. (2016). Histological and Histometric characterization of myocardial fibrosis in End-Stage hypertrophic cardiomyopathy: A Clinical-Pathological Study of 30 Explanted Hearts. Circ Heart Fail, Sep, 9(9). pii: e003090.
- Gersh, B. J., Maron, B. J., Bonow, R. O., et al. (2011). 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol, 58, e212-60.
- Gimeno, J. R., Tome-Esteban, M., Lofiego, C., et al. (2009). Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur Heart J, 30, 2599-2605.
- Girolami, F., Ho, C. Y., Semsarian, C., et al. (2010). Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol, 55(14), 1444-53
- Green, J. J., Berger, J. S., Kramer, C. M. & Salerno, M. (2012). Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging, 5, 370-377.
- Harris, K. M., Spirito, P., Maron, M. S., et al. (2006). Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation, 114, 216-25.
- Ho, C. Y., Abbasi, S. A., Neilan, T. G., et al. (2013). T1 measurements identify extracellular volume expansion in hypertrophic cardiomyopathy sarcomere mutation carriers with and without left ventricular hypertrophy. Circ Cardiovasc Imaging, 6, 415-422.
- Ho, C. Y., López, B., Coelho-Filho, O. R., et al. (2010). Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med, 363, 552-63.
- Ingles, J., Doolan, A., Chiu, C., et al. (2005). Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet, 42, e59.
- Ismail, T. F., Jabbour, A., Gulati, A., et al. (2014). Role of late gadolinium enhancement cardiovascular magnetic resonance in the risk stratification of hypertrophic cardiomyopathy. Heart,100, 1851-1858.
- Killu, A. M., Park, J. Y., Sara, J. D., et al. (2016). Cardiac resynchronization therapy in patients with end-stage hypertrophic cardiomyopathy. Europace, 0, 1-7.
- Kuck, K. H., Cappato, R., Siebels, J. & Ruppel, R. (2000). Randomized comparison of antiarrhythmic drug therapy with implantable defibrillators in patients resuscitated from cardiac arrest: the Cardiac Arrest Study Hamburg (CASH). Circulation, 102, 748-754.
- Lopes, L. R., Syrris, P., Guttmann, O. P., et al. (2015). Novel genotype–phenotype association demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart, 101, 294-301
- Maron, B. J., Haas, T. S., Shannon, K. M., et al. (2009). Long-term survival after cardiac arrest in hypertrophic cardiomyopathy. Heart Rhythm, 6, 993-7.
- Maron, B. J., Maron, M. S. & Semsarian, C. (2012). Double or compound sarcomere mutations in hypertrophic cardiomyopathy: a potential link to sudden death in the absence of conventional risk factors. Heart Rhythm, 9, 57-63.
- Maron, B. J., Rowin, E. J., Casey, S. A., et al. (2013). Risk stratification and outcome of patients with hypertrophic cardiomyopathy >= 60 years of age. Circulation, 127, 585-593.
- Maron, B. J., Rowin, E. J., Casey, S. A., et al. (2015). Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J Am Coll Cardiol, 65, 1915-28.
- Maron, M. S., Appelbaum, E., Harrigan, C. J., et al. (2008). Clinical profile and significance of delayed enhancement in hypertrophic cardiomyopathy. Circ Heart Fail, 1, 184-191.
- Maron, M. S., Finley, J. J., Bos, J. M., et al. (2008). Prevalence, clinical Significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation, 118, 1541-1549.
- Maron, M. S., Olivotto, I., Betocchi, S., et al. (2003). Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. New Engl J Med, 348, 295-303.
- Maron, M. S., Olivotto, I., Maron, B. J., et al. (2009). The case for myocardial ischemia in hypertrophic cardiomyopathy. J Am Coll Cardiol, 54, 866-75.
- Maron, M. S., Olivotto, I., Zenovich, A. G., et al. (2006). Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation, 114, 2232-2239.
- Maron, M. S., Rowin, E. J., Olivotto, I., et al. (2016). Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol, 67, 1399-409. Melacini, P., Basso, C., Angelini, A., et al. (2010). Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur Heart J, 31, 2111- 2123.
- Monserrat, L., Elliott, P. M., Gimeno, J. R., et al. (2003). Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol, 42, 873-879.
- Moon, J. C., Reed, E., Sheppard, M. N., et al. (2004). The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol, 43, 2260-4.
- O’Hanlon, R., Grasso, A., Roughton, M., et al. (2010). Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol 56: 867-874. O’Mahony, C., Jichi, F., Pavlou, M., et al. (2014). A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J, 35, 2010-201. O’Mahony, C., Lambiase, P. D., Quarta, G., et al. (2015). The long-term survival and the risks and benefits of implantable cardioverter defibrillators in patients with hypertrophic cardiomyopathy. Heart, 98, 116-25.
- O’Mahony, C., Tome-Esteban, M., Lambiase, P. D., et al. (2013). A validation study of the 2003 American College of Cardiology/European Society of Cardiology and 2011 American College of Cardiology Foundation/American Heart Association risk stratification and treatment algorithms for sudden cardiac death in patients with hypertrophic cardiomyopathy. Heart, 99, 534-541.
- Olivotto, I., Cecchi, F., Gistri, R., et al. (2006). Relevance of coronary microvascular flow impairment to long-term remodeling and systolic dysfunction in hypertrophic cardiomyopathy. J Am Coll Cardiol, 47, 1043-1048.
- Olivotto, I., Girolami, F., Sciagrà, R., et al. (2011). Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J Am Coll Cardiol, 58, 839-48.
- Oreto, Giuseppe., et al. (2009). L’elettrocardiogramma un mosaico a 12 tessere. Torino: Centro Scientifico Editore, 2009. Petersen, S. E., Jerosch-Herold, M., Hudsmith, L. E., et al. (2007). Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation, 115, 2418-2425.
- Prinz, C., Schwarz, M., Ilic, I., et al. (2013). Myocardial fibrosis severity on cardiac magnetic resonance imaging predicts sustained arrhythmic events in hypertrophic cardiomyopathy. Can J Cardiol, 29, 358-363.
- Quarta, C. C., Solomon, S. D., Uraizee, I., et al. (2014). Left Ventricular Structure and Function in Transthyretin-Related Versus Light-Chain Cardiac Amyloidosis. Circulation, 129, 1840-1849.
- Richard, P., Charron, P., Carrier, L., et al. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation, 107, 2227-2232.
- Rubinshtein, R., Glockner, J. F., Ommen, S. R., et al. (2010). Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail, 3, 51-58.
- Sadoul, N., Prasad, K., Elliott, P. M., et al. (1997). Prospective prognostic assessment of blood pressure response during exercise in patients with hypertrophic cardiomyopathy. Circulation, 96, 2987–2991.
- Semsarian, C., Ingles, J., Maron, M. S., et al. (2015). New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J Am Coll Cardiol, 65, 1249-54. Sorajja, P., Nishimura, R. A., Gersh, B. J., et al. (2009). Outcome of mildly symptomatic or asymptomatic obstructive hypertrophic cardiomyopathy: a long-term follow-up study. J Am Coll Cardiol, 54, 234-41.
- Spirito, P., Autore, C., Rapezzi, C., et al. (2009). Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation, 119, 1703-10.
- Spirito, P., Bellone, P., Harris, K. M., et al. (2000). Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med, 342, 1778-1785.
- Teekakirikul, P., Eminaga, S., Toka, O., et al. (2010). Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires TGF-β. J Clin Invest, 120, 3520-3529.
- Thaman, R., Gimeno, J. R., Murphy, R. T., et al. (2005). Prevalence and clinical significance of systolic impairment in hypertrophic cardiomyopathy. Heart, 91, 920- 925.
- The Antiarrhythmics versus Implantable Defibrillators (AVID) Investigators, (1997). A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. N Engl J Med, 337, 1576- 1583.
- Todiere, G., Aquaro, G. D., Piaggi, P., et al. (2012). Progression of myocardial fibrosis assessed with cardiac magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol, 60, 922-9.
- Veselka, J., Jensen, M. K., Liebregts, M., et al. (2016). Low procedure-related mortality achieved with alcohol septal ablation in European patients. Int J Cardiol, 209, 194- 95.
- Weissler-Snir, A., Adler, A., Williams, L., et al. (2016). Prevention of sudden death in hypertrophic cardiomyopathy: bridging the gaps in knowledge. Eur Heart J, Jul 1. pii: ehw268.


{kind=link}